Abstract
A subset of testicular sex cord-stromal tumors (SCST), which includes neoplasms with mixed histology, cannot be classified into a specific histologic subtype. This study evaluated the clinicopathologic, immunophenotypic and molecular features of 26 SCST not amenable to specific classification by expert uropathologists. Median age at diagnosis was 43 years and median tumor size was 2.4 cm. Follow-up information was available for 18 (69%) patients, with evidence of an aggressive clinical course in 6 patients (4 alive with disease, 2 dead of disease 3 months and 6 months after orchiectomy). Microscopically, SCST not amenable to specific classification demonstrated monophasic epithelioid (9/26, 35%), monophasic spindle cell (5/26, 19%), and biphasic or mixed histology (12/26, 46%). One or more aggressive histopathologic features were seen in 11 cases. DNA sequencing was successful in 22 tumors. Pathogenic CTNNB1 and APC alterations were seen in 7 (33%) and 2 (10%) cases, respectively, with additional variants (e.g., CDKN2A, RB1, TP53, BRCA2) being identified in individual cases. Combined evaluation of morphology, sequencing data and beta-catenin immunohistochemistry resulted in reclassification of 6 (23%) tumors as Sertoli cell tumor, not otherwise specified. This was supported by comparing the methylation profiles of a subset of these tumors and those of typical Sertoli cell tumors. Additionally, a subset of 5 neoplasms (19%) with spindle cell or biphasic histology and SMA expression was characterized by hyperdiploid genomes with recurrent chromosomal gains and absence of driver mutations, possibly representing a distinct tumor type. The SCST that remained not amenable to specific histologic classification (15/26, 58%) were enriched for aggressive histologic features and malignant clinical behavior. In conclusion, this study demonstrated that a subset of testicular SCST that were originally not amenable to specific classification could be reclassified by combined evaluation of morphology, immunohistochemistry and molecular data.
Similar content being viewed by others
Introduction
Sex cord-stromal tumors (SCST) constitute <5% of all testicular neoplasms in adults and up to 8% in pediatric patients1, and often represent a diagnostic challenge due to their varied clinicopathologic characteristics. Small subsets of these tumors that is not amenable to specific subclassification by morphology and immunohistochemistry are currently grouped under one of two categories in the World Health Organization (WHO) classification of genitourinary tumors: SCST, not otherwise specified (NOS) and mixed SCST2. These categories include neoplasms with diverse growth patterns, cytomorphology and clinical behavior. A subgroup is characterized by the presence of neoplastic sex cord-stromal elements and entrapped non-neoplastic germ cells3. Testicular SCST with a combination of Sertoli and Leydig cells are vanishingly rare and considered a type of mixed SCST4. SCST with predominantly spindle cells and co-expression of smooth muscle actin (SMA) and S100 protein5 are now considered a distinct entity designated myoid gonadal stromal tumor (MGST)6.
Recent molecular analyses have identified alterations that are characteristic (albeit often not pathognomonic) of certain histologic subtypes of SCST and may aid with subclassification7,8,9,10. Therefore, we hypothesized that some SCST, NOS and mixed SCST could be reclassified by a combined evaluation of morphologic, immunohistochemical and molecular data. Prior gene-specific analyses of unclassified ovarian SCST demonstrated that a subset can be reclassified using canonical molecular alterations such as the FOXL2 p.C134W hotspot mutation seen in >90% of ovarian adult granulosa cell tumors11. This mutation is rare but does occur occasionally in testicular adult granulosa cell tumor12,13, a tumor type that demonstrates morphologic overlap with SCST, NOS. In this study we performed an integrated evaluation of the clinicopathologic, immunophenotypic and molecular features of a multi-institutional series of testicular SCST that could not be originally classified into specific histologic subtypes.
Materials and methods
This study was performed with approval of the Institutional Review Boards of Brigham and Women’s Hospital (MGB Insight 4.0, protocol #2021P002289) and the other participating institutions (when applicable).
Identification and accrual of cases
The pathology databases of the participating institutions and personal consultation files of the authors were queried for cases diagnosed as “unclassified” SCST or SCST, NOS/mixed SCST in male patients. Cases with archival formalin-fixed paraffin-embedded material (FFPE, blocks or slides) available for molecular analysis were included in the study. Slides were retrieved and reviewed at the participating institutions, followed by re-review of selected slides at BWH (SS and AMA). Of note, spindle cell tumors with storiform and/or fascicular growth patterns and unequivocal co-expression of SMA and S100 protein were re-classified as MGST and excluded from the study.
Clinical data and pathology review
Clinical and demographic data were obtained from electronic medical records, pathology reports and consultation letters. The following information was collected for each patient: age at diagnosis, tumor size, tumor site, results of relevant immunohistochemical stains (performed as part of the initial diagnostic workup), and follow-up data (including recurrences and death). The following histopathologic features were evaluated: cytomorphology, architecture, number of mitoses per 10 high power fields (HPF), overall cellularity, pleomorphism, infiltrative growth, necrosis, and lymphovascular invasion. Cytomorphology was defined as monophasic epithelioid (>90% epithelioid cells), monophasic spindle cell (>90% spindle cells), and biphasic or mixed. Biphasic histology was defined by the absence of a dominant cytomorphology (any cytomorphology representing <90% of the cellularity), with different combinations of spindled, stellate, epithelioid and histiocytoid cells. Mixed histology was defined by the presence of two distinct lines of differentiation/cell types (e.g., Sertoli cells and Leydig cells). Pleomorphism was subjectively evaluated as minimal/moderate when there was mild-to-moderate nuclear atypia and variation of nuclear size between neoplastic cells and severe when there was marked nuclear atypia and pronounced variation in nuclear size (including giant tumor cells). Tumors were assessed for the presence of aggressive features, including >5 mitoses per 10 HPF, size ≥5.0 cm, severe nuclear pleomorphism, necrosis, and lymphovascular invasion1,6. Tumors associated with recurrence, metastasis, or death due to disease were considered clinically malignant.
Massively parallel sequencing (OncoPanel)
A 447-gene targeted solid tumor next-generation sequencing panel (OncoPanel) was performed as previously described14,15. Briefly, fresh frozen paraffin-embedded (FFPE) tissue was manually dissected from unstained histology slides previously marked by a pathologist (SS and AMA) to attempt to obtain a neoplastic cellularity ≥20%. DNA was extracted using a commercially available kit (Qiagen, Valencia, CA) according to the manufacturer’s recommendations, and DNA was subsequently fragmented by sonication. A target input of 200 ng of DNA per sample was used, with a minimum threshold of 100 ng for sample acceptance. Sequencing libraries were prepared with the TruSeq LT library preparation kit (Illumina, San Diego, California) and target sequences were selected by hybridization (DNA probes custom-designed by Agilent SureSelect; Agilent Technologies, Santa Clara, CA). After sequencing by synthesis on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA), deconvolution of multiplexed samples, read alignment, variant calling and annotation was performed using a clinically validated informatics pipeline14,15,16. In-house algorithms were used to identify mutational signatures (POLE, APOBEC, smoking, UV) and mismatch repair status17,18. Events present at a frequency >0.1% in the gnomAD database (Broad Institute) were filtered out to avoid contamination with germline variants. Loss of heterozygosity and copy number alterations were evaluated using the log (2) mean read ratio and variant allele frequency (VAF)19. The reported variants were reviewed and tiered for actionability and biological relevance by two independent molecular pathologists (LMS and HKT).
Immunohistochemistry
Immunohistochemistry (IHC) was performed with primary antibodies against beta-catenin (clone 14, mouse monoclonal, BD Biosciences, San Jose, CA; dilution 1:2000). Nuclear expression of beta-catenin (with or without concurrent cytoplasmic expression) in >5% of the tumor cells was considered a positive result. Positive and negative controls were run and assessed in parallel.
Methylation analysis
Four study cases (cases 3, 6, 12 and 24) with pathogenic CTNNB1 or APC variants, nonfocal nuclear beta-catenin expression and sufficient FFPE material available underwent DNA methylation analysis. Additionally, the DNA methylation profile of 6 Sertoli cell tumors, NOS of unknown mutational status with typical histology and diffuse nuclear beta-catenin expression was assessed for comparison. Whole genome DNA methylation profiling was performed as previously reported20. In brief, FFPE tissue was dissected to enrich for tumor cells before DNA extraction. The DNA was processed for hybridization and fluorescence staining on the Infinium MethylationEPIC (850 k) BeadChip array (Illumina, San Diego, USA) according to the manufacturer’s instructions for FFPE samples. Arrays were scanned in an Illumina iScan microarray scanner, and the generated raw idat files were analyzed in R (R Foundation for Statistical Computing, Vienna, Austria). Raw intensities were processed with Bioconductor R package Minfi21. Each sample was assessed for quality by mean detection p-value (p < 0.05). All samples (n = 10) passed quality control metrics. Each sample was normalized by quantile normalization, followed by the removal of probes that failed in one or more samples (detection p < 0.01), and the removal of probes with single-nucleotide polymorphisms. Beta-values were calculated with the default offset value 100, recommended for Illumina assays. Differential methylation of typical Sertoli cell tumor and mixed SCST/SCST, NOS samples was assessed. After fitting linear models with limma (3.48.3), top differentially methylated probes were selected based on adjusted p-values (p < 0.05). Hypomethylation was defined as a Beta value <0.2, whereas hypermethylation was defined as a Beta value >0.8. A heatmap was generated by ComplexHeatmap (2.8.0) based on the hierarchical clustering of the top 1000 differentially methylated probes22.
Results
Clinicopathologic and immunophenotypic features of sex cord-stromal tumors not amenable to histologic subclassification
After excluding 2 lesions that were reclassified as myoid gonadal stromal tumor, 26 testicular SCST not amenable to specific histologic subclassification were included in this study. These cases were originally diagnosed between 1995 and 2021 at multiple institutions. The median patient age at presentation was 43 years (range: 2 mo–72 yrs). All specimens were resections (i.e., radical or partial orchiectomies), including a case with a paired lymph node metastasis that was also reviewed for the study. Median tumor size was 2.4 cm (range: 0.6 cm–9.8 cm). Histologic review demonstrated marked heterogeneity, with variable architectural patterns and cytomorphology (Table 1). Tumors were generally well-circumscribed but unencapsulated, with only 4 tumors demonstrating infiltrative growth into adjacent structures (e.g., hilar soft tissue) in the slides reviewed. Lymphovascular invasion and necrosis were present in 3 tumors each. Mitotic activity ranged from <1 mitosis per 10 HPF to 169 mitoses per 10 HPF (median 3 mitoses per 10 HPF). Severe pleomorphism was noted in 3 tumors, whereas the rest exhibited only minimal or moderate pleomorphism. Overall, 11 tumors (42%) had at least one aggressive histologic feature, and 5 of these tumors (19%) had ≥2 features (i.e., aggressive histology). Median patient age for tumors with aggressive features was 54 years old (range: 5 yrs – 68 yrs), compared with 42 years old for non-aggressive tumors (range: 2 mo–72 yrs).
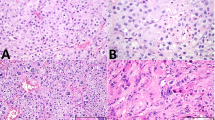
Monophasic epithelioid, monophasic spindle cell, and biphasic or mixed histology were identified in 9, 5 and 12 cases, respectively. Monophasic epithelioid tumors exhibited multiple growth patterns, including solid sheets or nests, fascicles, trabeculae, and cords (Fig. 1A). Spindle cell-predominant tumors demonstrated fascicular, whorled and/or sheet-like growth patterns (Fig. 1B). As mentioned above, the biphasic/mixed category included tumors with multiple cytomorphologies (n = 8) or multiple cell types (i.e., multiple lines of differentiation; n = 2). The former group comprised tumors with mixed populations of spindle, stellate, epithelioid and/or histiocytoid cells (Fig. 1C, D). In contrast, the latter group included neoplasms with a combination of Sertoli and Leydig cells.

A Malignant sex cord-stromal tumor with monophasic epithelioid morphology (case 12). B Sex cord-stromal tumor with monophasic spindle cell morphology (case 8). C Biphasic sex cord-stromal tumor with epithelioid (right) and spindle cells (case 5). D Biphasic sex cord-stromal tumor (case 3) with epithelioid and spindle cells (inset). Both components of this lesion demonstrated diffuse nuclear beta-catenin expression (not shown).
Information on the immunohistochemistry performed as part of the original diagnostic evaluation was available for 18 cases. Tumors were frequently positive for SF1 (14/18), inhibin (13/16) and calretinin (8/13). Expression of keratins and WT1 was also seen in a subset of cases (5/18 and 4/8, respectively). Only 2/10 tumors demonstrated weak and focal (i.e., noncontributory) expression of S100 protein. Germ cell markers including SALL4 and OCT3/4 were negative in all cases in which they were performed. Beta-catenin immunohistochemistry was performed de-novo for this study on all cases with additional FFPE material available (n = 20). Of these, 8 cases (40%, cases 2-6, 12, 16 and 24) demonstrated nonfocal nuclear staining, with diffuse staining (i.e, 80-100% of the tumor cells) in 5 of them. Among cases with nuclear beta-catenin expression, 4 were predominantly epithelioid and 4 were biphasic or mixed. Importantly, 2 cases that comprised a mixed population of Sertoli and Leydig cells (cases 2 and 6) demonstrated nuclear beta-catenin expression exclusively in Sertoli cells. Case 1, which harbored a gain-of-function CTNNB1 mutation (see below), did not have additional FFPE tissue for beta-catenin immunohistochemistry.
DNA sequencing
Twenty-two of the 26 cases passed standard quality assurance metrics. Tumor mutational burden was generally low across the cohort (median 3.80 mutations per megabase). All cases were mismatch repair-proficient, and no specific signatures were identified.
Pathogenic SNVs and/or indels predicted to be relevant for tumorigenesis [variant allele frequency (VAF) sufficient for clonal or subclonal event, based on heterozygosity or homozygosity at locus] were identified in 9 cases (Fig. 2). All these cases harbored pathogenic mutations in genes encoding WNT signaling proteins, including 7 gain-of-function CTNNB1 alterations and 2 inactivating APC mutations with evidence of loss of heterozygosity (LOH). CTNNB1 variants constituted the most frequent recurrent molecular alteration. One of the tumors (case 5) harbored an in-frame CTNNB1 deletion that disrupted a GSK3β phosphorylation site, making the protein resistant to degradation23 and available for activation of WNT signaling24. Other variants previously reported in specific types of SCST were not identified (e.g., FOXL2 p.C134W, AKT internal tandem duplications, and mutations in PRKAR1A, FH, and DICER1). Loss-of-function TP53 and CDKN2A mutations with evidence of LOH were seen in one case each, and they were both present in the context of concurrent WNT pathway gene mutations. Non-recurrent pathogenic SNVs included CHEK2, KMT2D, PMS2, ARID1A, SMARCB1, MSH6, SETD2 and TET2; however, these alterations either exhibited low VAFs or retained heterozygosity and were not considered major contributors to oncogenesis. One case harbored a BRCA2 rearrangement expected to result in loss of protein function (case 19). Additional large structural variants were not identified with this sequencing panel.

SVs structural variants, CNVs copy number variants, Histo histology, MGST myoid gonadal stromal tumor, NGS next generation sequencing, CN-LOH copy-neutral loss of heterozygosity, TST testicular stromal tumor. “Aggressive Histo.” denotes cases with at least one of the following findings: >5 mitoses per 10 HPF, size ≥5.0 cm, severe nuclear pleomorphism, necrosis, and lymphovascular invasion. Reclassification: S Sertoli cell tumor, NOS.
Arm-level and chromosome-level CNVs were identified in 13 tumors (Fig. 2). Numerous chromosome-level copy number gains consistent with hyperdiploidy were identified in 6 tumors (cases 7–11 and 16), including recurrent gains of chromosomes 3, 6, 7, 8, 12, 15, 17, and 20. Notably, most of these hyperdiploid tumors (5/6, cases 7–11) did not harbor concurrent SNVs considered relevant for oncogenesis. These cases included 3 monophasic spindle cell tumors and 2 biphasic tumors (see re-review of cases below). The one case with concurrent oncogenic variants (case 16) harbored a gain-of-function CTNNB1 indel and a loss-of-function CDKN2A mutation with LOH. This tumor exhibited multiple aggressive pathologic features and was associated with a malignant clinical course. Three additional cases (cases 12, 18 and 24) had evidence of genome-wide instability with multiple arm-level and chromosome-level copy number changes. One of these cases also exhibited biallelic loss of RB1 and KDM6A (case 18), while another showed biallelic loss of RB1 and genome-wide loss of heterozygosity (copy neutral-LOH for all chromosomes except chromosome 21, case 12). Other CNVs included focal amplification of CRKL (estimated 7 copies) with concurrent -1p, -6p and a focal deletion within 22q (case 4, focal deletion not shown in the figure), chromosome 15 triploidy (case 23), and isolated 3q arm gain (case 3).
Re-review of cases with WNT pathway activation and hyperdiploid genomes
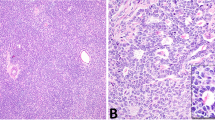
We next sought to reclassify tumors based on the integrated evaluation of morphology, IHC and DNA sequencing data. Beta-catenin IHC was performed on all cases with additional FFPE material available, including 8/9 cases with mutations in WNT pathway genes (cases 2–6, 12, 16, and 24), all of which exhibited nuclear beta-catenin expression in at least a subset of the neoplastic cells. Case 1, which harbored an activating CTNNB1 mutation, had no additional FFPE material available for beta-catenin immunohistochemistry. Re-review of cases with nonfocal nuclear beta-catenin expression and/or molecular evidence of WNT pathway activation as a driver event identified 4 tumors (cases1, 3, 4, and 5; Fig. 3) with areas that imperfectly mimicked growth patterns seen in Sertoli cell tumors, NOS. More specifically, these cases had foci suggestive of tubular and/or retiform architecture. One of these cases had a predominant spindled component, with foci that resembled solid tubules/cords (case 5, “Sertoli-stromal” tumor). Two additional cases (cases 2 and 6) had a mixture of Sertoli and Leydig cells, with nuclear beta-catenin expression restricted to the Sertoli cell component, suggesting that the Leydig cells were non-neoplastic. In context, these 6 cases could be reclassified as Sertoli cell tumors with unusual histologic features (cases 1-6), while the remaining cases with WNT pathway activation remained unclassifiable (cases 12, 16 and 24) (Fig. 3A–D and Supplementary Figure 1).

A Case 4 exhibited predominantly solid sheets and nests of epithelioid cells. However, there were small foci reminiscent of the retiform growth pattern seen in Sertoli cell tumors, not otherwise specified (inset). Foci reminiscent of tubular architecture were also present (not shown) B Case 4 demonstrated diffuse nuclear expression of beta-catenin and harbored a CTNNB1 mutation and was reclassified as a Sertoli cell tumor, not otherwise specified. C Case 2 comprised an even mixture of Sertoli and Leydig cells. D The Sertoli cell component demonstrated diffuse nuclear expression of beta-catenin. The absence of nuclear beta-catenin expression in the Leydig cells suggests that, albeit abundant, they are most likely non-neoplastic. This tumor harbored an APC variant and was reclassified as Sertoli cell tumor, not otherwise specified.
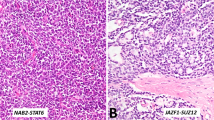
Next, we considered the cohort of 6 tumors with hyperdiploid genomes and recurrent chromosome-level gains (cases 7–11 and 16). Case 16, a clinically malignant tumor, had concurrent pathogenic CTNNB1 and CDKN2A variants, whereas the remaining 5 tumors had no evidence of concurrent driver mutations. Three of the latter (cases 7–9) were monophasic spindle cells lesions, while the remaining two (cases 10 and 11) had a biphasic morphology, with both epithelioid and spindle cells. Case 10 had a predominant spindle cell population (~80%), with a smaller component of epithelioid cells arranged in cords/solid tubules. In both of these biphasic cases, there was a smooth transition between the epithelioid and spindle cells. These 5 tumors were remarkably homogeneous molecularly, shared multiple morphologic features, lacked worrisome histologic findings, consistently expressed SMA and demonstrated a nonaggressive clinical course. Given these commonalities, we believe that these 5 neoplasms may represent a distinct tumor type (Fig. 4). Like myoid gonadal stromal tumor, they consist predominantly of bland spindle cells that grow in fascicular or storiform patterns entrapping seminiferous tubules. However, unlike myoid gonadal stromal tumor, these lesions do not express S100 and a subset exhibits biphasic morphology with a component of epithelioid cells arranged in cords, tubules and/or trabeculae (i.e., sex cord component; Table 2). Given the morphologic resemblance to myoid gonadal stromal tumor, we used the term “myoid gonadal stromal tumor-like tumor” to designate these neoplasms. Of note, the 2 tumors that were initially reclassified as myoid gonadal stromal tumor and excluded from the series also underwent DNA sequencing. One of them harbored a frameshift ATR variant at a VAF of 73% and a likely germline BRIP1 splice site variant but copy number analysis could not be performed due to a wide signal dispersion (i.e., noise). The remaining myoid gonadal stromal tumor had no somatic SNVs and its number copy profile was characterized by chromosome-level copy number gains involving chromosomes 3, 6, 8, 9, 11 and 12.

Cases 7 (A), 8 (B) and 9 (C) exhibited a monophasic spindle cell morphology with a predominantly fascicular architecture and occasional whorls. Case 10 demonstrated a biphasic architecture with fascicles of spindle cells and epithelioid cells arranged in solid tubules/cords (D, upper right). There was a smooth transition between the two components of this tumor (E). F Case 11 also had a biphasic architecture, with a mixture of spindle and epithelioid cells, with a smooth transition between them.
In total, 6 tumors were reclassified as Sertoli cell tumor, NOS with unusual morphologic features and 5 were interpreted as a distinct tumor type by combined evaluation of morphology, immunohistochemistry and sequencing data. None of these 11 tumors exhibited >2 aggressive pathologic features (as defined in Materials and Methods) or a malignant clinical behavior, but case 4 (reclassified as Sertoli cell tumor) measured 4.6 cm and demonstrated 11 mitoses per 10 HPF. Subtraction of these cases yielded a cohort of 15 SCST not amenable to classification, including all tumors with a malignant clinical course (6/6, 100%) and most tumors with aggressive histologic features (10/11, 91%).
DNA Methylation Analysis
Methylation analysis was performed to compare the methylation profiles of 4 SCST not amenable to specific histologic sub-classification and 6 Sertoli cell tumors. The study cases were selected based on the presence of molecular evidence of WNT pathway activation, nonfocal nuclear beta-catenin expression by IHC and the availability of additional FFPE tissue (cases 3, 6, 12 and 24). The comparator cases (Sertoli cell tumors, NOS) were selected based on the presence of typical histomorphology and diffuse nuclear beta-catenin expression by IHC. Hierarchical clustering based on the top 1000 differentially methylated probes demonstrated that the methylation profiles of 2 SCST not amenable to specific histologic classification (cases 3 and 6) were remarkably similar to those of typical Sertoli cell tumors (i.e., they clustered together with Sertoli cell tumor, NOS). The remaining 2 SCST not amenable to histologic classification (cases 12 and 24) had clearly different methylation profiles and clustered separately (Fig. 5). The two SCST not amenable to histologic classification that clustered with the Sertoli cell tumors (cases 3 and 6) harbored gain-of-function CTNNB1 mutations as relatively isolated findings (VAF~cellularity/2) and lacked aggressive histologic features or a malignant clinical course. These two neoplasms had been reclassified as Sertoli cell tumor, NOS based on combined evaluation of morphology, immunohistochemistry and sequencing data. By comparison, the two SCST not amenable to specific histologic classification that formed a distinct separate cluster (cases 12 and 24) demonstrated numerous additional molecular alterations including mutations and widespread copy number alterations, and they both exhibited aggressive histologic features and a malignant clinical course. Neither of these two neoplasms had been reclassified by combined assessment of morphology, immunohistochemistry and sequencing data.

Hierarchical clustering using top 1000 differentially methylated probes for 4 sex cord-stromal tumors not amenable to specific histologic classification (SCST, NOS or mixed SCST) and 6 Sertoli cell tumors shows two tumors (study cases 12 and 24) forming an independent cluster while two tumors (study cases 3 and 6) cluster with Sertoli cell tumors. SCST sex cord-stromal tumor, NOS not otherwise specified. The asterisks (*) designate six individual Sertoli cell tumors with typical histologic features and diffuse expression of beta-catenin by immunohistochemistry. The numbers in the green boxes designate the study cases. The methylation scores are in Beta-values (see Materials and Methods section). The micrographs illustrate the two study cases (cases 12 and 24) that clustered independently.
Correlation of histologic, molecular and outcome data
Follow-up information was available for 18 cases (median 9 mo, range: 1 mo–25 yrs; cases 1, 2, 5-7, 9, 11-16, 18, 20, 21, 23, 24 and 26). Of the cases with follow-up, 12 were alive with no evidence of disease, while 6 exhibited a malignant clinical course. This included two patients with local recurrences (9 mo and 4 yrs), two patients with metastatic disease (5 mo and 9 mo), and two patients who were dead of disease (3 mo and 6 mo) (Table 1). Of note, none of the cases reclassified as Sertoli cell tumor or interpreted as a distinct stromal tumor type (myoid gonadal stromal tumor-like tumor) had a malignant clinical course. All cases with a malignant clinical behavior were predominantly epithelioid and had at least one aggressive histologic feature (6/6, 100%), with most (4/6, 67%) demonstrating ≥2 aggressive histologic features (Table 1). Likewise, the majority (6/8, 75%) of tumors with at least one aggressive histologic feature and known follow-up demonstrated a malignant clinical course (recurrence, metastasis, and/or death of disease).
Among cases with malignant behavior, case 12 had concurrent biallelic APC inactivation, biallelic RB1 inactivation and multiple copy number changes with widespread LOH; case 16 had an activating CTNNB1 variant with concurrent biallelic loss of CDKN2A and multiple chromosomal gains; case 18 had an inactivating BRCA1 rearrangement with concurrent biallelic RB1 loss and multiple chromosome-level losses; and case 24 had an activating CTNNB1 mutation with multiple concurrent chromosome-level losses. Case 21 had no molecular findings; however, cellularity in this case was well below 20% due to the presence of a prominent neutrophilic infiltrate. Case 13, which was also malignant, failed sequencing.
Discussion
Testicular SCST constitute ~5% of all testicular neoplasms and include Leydig cell tumor and Sertoli cell tumor, NOS, as well as other defined histologic subtypes that are relatively rare25. Subsets of testicular SCST exhibit morphologic and immunophenotypic features that preclude their classification into known histologic subtypes7. These neoplasms, which were previously called “unclassified” SCST, are now termed either ‘SCST, NOS’ or ‘mixed SCST’2.
Most testicular SCST are clinically indolent, but a subset exhibits malignant behavior with poor response to systemic treatment26,27. Presently, there are no unified criteria to predict malignant potential across the different types of testicular SCST. In adult granulosa cell tumors, the only feature consistently associated with malignancy is tumor size ≥5 cm28. In early series of Leydig cell tumors, the presence of different combinations of the following features was associated with malignancy: tumor size ≥5.0 cm, >3 mitoses per 10 high-power fields, vascular invasion, infiltrative growth, cytologic atypia, or necrosis1. A recent meta-analysis has suggested that Leydig cell tumors larger than 3 cm with any additional adverse histopathologic feature (e.g., tumor necrosis) may have significant malignant potential, especially in adult patients29. Early series of Sertoli cell tumor, NOS demonstrated that the presence of two or more of the following features was associated with malignant cases: tumor size >5 cm, moderate or severe nuclear atypia, >5 mitoses per 10 HPF, vascular invasion and tumor necrosis30. A recent meta-analysis has suggested that a size larger than 2.4 cm, patient age >27.5 years and the presence of adverse histopathologic features might predict aggressive behavior in Sertoli cell tumor, NOS31. Up to 20% of SCST that cannot be classified as one of the known histologic subtypes exhibit malignant behavior32. The WHO classification of genitourinary tumors recommends adaptation of the features used to predict aggressive behavior in Sertoli and Leydig cell tumors to all SCST2, although it is still uncertain whether these criteria can be extrapolated from one SCST type to another.
In recent years, the molecular features of several histologic types of testicular SCST have been described. The overwhelming majority of Sertoli cell tumors, NOS harbor gain-of-function CTNNB1 variants that activate WNT signaling9,33,34. The presence of these variants correlate with strong and diffuse nuclear and cytoplasmic expression of beta-catenin in the neoplastic cells9. Subsets of Leydig cell tumors harbor inactivating FH variants, gain-of-function CTNNB1 variants present at VAFs suggestive of subclonal events8 and MDM2 amplifications35,36. Most large cell calcifying Sertoli cell tumors harbor inactivating PRKAR1A variants and demonstrate loss of PRKAR1A protein expression by IHC37. A comprehensive assessment of the morphologic, immunophenotypic and molecular features of SCST not amenable to specific histologic classification had not been undertaken previously. We hypothesized that some of these neoplasms may harbor genetic variants that, in combination with morphologic and immunohistochemical findings, may aid in their classification.
Reclassification of cases as Sertoli cell tumor
In this study, combined evaluation of morphology, IHC and sequencing data allowed reclassification of 6 tumors as Sertoli cell tumor. The lesions comprised in this group were mixed neoplasms with a combination of Sertoli and Leydig cells, as well as tumors with predominantly biphasic morphology including foci with spindle or stellate cells and epithelioid or histiocytoid cells. Only 1 case was a purely epithelioid tumor (case 4) measuring 4.6 cm, with a largely solid growth pattern and up to 11 mitoses per 10 HPF. All of the tumors that could be reclassified had small foci that were suggestive of tubular and/or rete-like architecture, although well-formed tubules were largely absent in all cases except for case 2 (mixed tumor with Sertoli and Leydig cells). Of note, all cases that could be reclassified exhibited nonfocal nuclear beta-catenin expression and/or harbored CTNNB1 or APC variants consistent with a clonal gain-of-function event or biallelic inactivation, respectively.
DNA methylation signatures in cancer are determined by cell of origin/line of differentiation as well as driver mutations, and can be used to classify tumors that are challenging to differentiate based on morphology and immunohistochemistry38,39,40. We performed hierarchical clustering based on methylation profile in 6 Sertoli cell tumors, NOS, and 4 SCST not amenable to specific classification with nonfocal nuclear beta-catenin expression. The latter included three tumors with CTNNB1 mutations (cases 3, 6 and 24) and one with biallelic APC inactivation (case 12). Clustering supported our reclassification of 2 cases as Sertoli cell tumor, NOS with unusual growth patterns (cases 3 and 6). The remaining two cases that clustered independently (cases 12 and 24) could not be reclassified as Sertoli cell tumor based on combined assessment of morphology, beta-catenin IHC and DNA sequencing data. An important point highlighted by the methylation data is that WNT pathway activation (by means of gain-of-function CTNNB1 or loss-of-function APC variants) is not in and of itself diagnostic of Sertoli cell tumor, NOS, especially in neoplasms that exhibit aggressive histologic features and nondescript morphology. In contrast, tumors with evidence of WNT pathway activation, foci reminiscent (but not diagnostic) of Sertoli cell tumor and absence of worrisome histologic findings can be classified as Sertoli cell tumor, NOS. More specifically, morphologically bland cases with solid, reticular or nondescript growth patterns and foci that imperfectly mimic tubular or retiform architecture can be classified as Sertoli cell tumor if they harbor activating CTNNB1 variants present VAFs consistent with a clonal gain-of-function event, biallelic APC inactivation and/or exhibit nonfocal nuclear beta-catenin expression. Tumors with a mixed population of Sertoli and Leydig cells with nonfocal nuclear beta-catenin staining restricted to the former component can also be classified as Sertoli cell tumors. This suggests that, at least in a subset of testicular SCST with mixed populations of Sertoli and Leydig cells, the latter component is nonneoplastic. This contrasts with ovarian Sertoli-Leydig cell tumor, which is characterized by DICER1 mutations4,41. In our study, DICER1 alterations were not identified.
Identification of a purportedly distinct testicular stromal tumor type (myoid gonadal stromal tumor-like tumor)
Five tumors with monophasic spindle cell or biphasic histology (cases 7-11) harbored copy number changes consistent with a hyperdiploid genome, including recurrent gains of chromosomes 3, 6, 7, 8, 12, 15, 17, and 20, without concurrent oncogenic mutations. These gains are similar to those previously reported in an “unclassified” infantile SCST42. All of these tumors were clinically benign and expressed SMA. Three of these tumors were exclusively spindled (cases 7-9), with the remaining 2 (cases 10–11) demonstrating a biphasic population of epithelioid and spindled cells, with a smooth transition between them. Case 10 was predominantly spindled, with scattered foci of epithelioid cells arranged in solid tubules or cords. Based on their remarkably similar molecular profile and the presence of shared morphologic features, these neoplasms may constitute a distinct testicular stromal tumor type. Additional studies are required to further characterize these lesions and assess whether they represent part of the spectrum of myoid gonadal stromal tumor.
Sex cord-stromal tumors that remained not amendable to specific classification
In this study, 15 cases remained not amenable to specific classification after combined evaluation of morphology, IHC and sequencing results. Importantly, 3 tumors with WNT pathway activation and/or diffuse nuclear beta-catenin expression that could not be reclassified had aggressive histologic features and a malignant clinical course (cases 12, 16 and 24). This highlights that, among SCST, CTNNB1 and APC mutations are not pathognomonic of Sertoli cell tumor. For instance, CTNNB1 mutations are usually present in Leydig cell tumors, albeit often at VAFs consistent with a subclonal event8. The identification of APC mutations with LOH in this study suggests that some SCST may arise in patients with germline APC variants. However, confirmatory clinical and germline data was not available.
Notably, all tumors with malignant clinical behavior (12, 13, 16, 18, 21, and 24) remained unclassified after molecular analysis. Accordingly, SCST that remained not amenable to specific histologic classification were enriched for cases with at least one aggressive histologic feature (10/15, 67%), monophasic epithelioid morphology (8/15, 53%) and a malignant clinical behavior (6/15, 40%). Given the biological and clinical heterogeneity of SCST not amenable to specific histologic classification, a single designation seems ill-fitted for these neoplasms. We believe that “poorly-differentiated SCST” or “undifferentiated SCST” are likely better terms than “SCST, NOS” for neoplasms with multiple aggressive histologic and/or clinical features.
Besides CTNNB1 and APC variants, other molecular alterations identified in the series were heterogeneous, but potentially useful to explain tumorigenesis and guide treatment in individual patients. For example, one malignant tumor with aggressive histologic features harbored a pathogenic CDKN2A mutation with loss of heterozygosity (case 16). Interestingly, this tumor was hyperdiploid, and demonstrated a concurrent CTNNB1 mutation with VAFs suggesting that CTNNB1 and CDKN2A alterations were present in all tumor cells. Similar mutations have previously been reported in metastatic SCST of the testis, including undifferentiated tumors and Sertoli cell tumors36 Another tumor with epithelioid morphology and prominent mitotic activity harbored a potentially inactivating BRCA2 rearrangement (case 18). In agreement with a functional loss of the BRCA2 protein, this tumor exhibited genome-wide chromosomal instability including homozygous copy-loss of KMD6A and RB1. These alterations could be used to direct targeted therapy including CDK4/CDK6 inhibition, PARP inhibition and AURKA inhibition.
This study has several limitations. First, the series includes tumors with heterogeneous morphologic features. This is somewhat unavoidable, since SCST, NOS and mixed SCST are diagnoses of exclusion. Moreover, this study comprised “unclassified” SCST diagnosed or seen in consultation by expert uropathologists from multiple institutions, likely capturing the spectrum of SCST, NOS and mixed SCST seen in most large practices. Second, a targeted sequencing panel was used to evaluate the cases, limiting the detection of novel variants. However, we decided to use our institutional sequencing panel because it is a clinically validated tool with known performance characteristics that has demonstrated excellent reliability over the years for the evaluation of clinical and research cases. Hence, the variants identified by this platform are most likely biologically relevant. Third, germline data was not available to determine the origin of some variants (e.g., APC mutations). Finally, follow up data were limited for most patients. The last two limitations were difficult to circumvent, because most cases included in the study were seen in consultation and had limited clinical information and archival material available.
In conclusion, this study demonstrates that combined evaluation of morphology, IHC and molecular data may help reclassify a significant subset of SCST that would otherwise remain unclassified. More specifically, SCST with bland histology (including a subset with mixed populations of Sertoli and Leydig cells), molecular evidence of WNT pathway activation and/or diffuse nuclear beta-catenin expression and growth patterns reminiscent (but not diagnostic) of Sertoli cell tumor, NOS can likely be classified as such. Finally, a group of SCST with spindle cell or biphasic morphology, SMA expression and hyperdiploid genomes may represent a distinct testicular sex cord stromal tumor type. After excluding these reclassifiable and purportedly novel neoplasms, the SCST that remained not amenable to specific histologic classification were enriched for aggressive histologic and clinical features.
Data availability
The data generated in this study are available from the corresponding author upon reasonable request.
References
Kim, I, Young, RH, Scully, RE. Leydig cell tumors of the testis. A clinicopathological analysis of 40 cases and review of the literature. Am J Surg Pathol 9, 177–92 (1985).
WHO Classification of Tumours Editorial Board. Urinary and male genital tumours [Internet], 5th edn. International Agency for Research on Cancer: Lyon (France), 2022, [cited 2022-05-01]. Available from: https://tumourclassification.iarc.who.int/chapters/36.
Ulbright, TM, Srigley, JR, Reuter, VE, Wojno, K, Roth, LM, Young, RH. Sex cord-stromal tumors of the testis with entrapped germ cells: a lesion mimicking unclassified mixed germ cell sex cord-stromal tumors. Am J Surg Pathol 24, 535–42 (2000).
Young, RH. Sex cord-stromal tumors of the ovary and testis: their similarities and differences with consideration of selected problems. Mod Pathol 18 Suppl 2, S81-98 (2005).
Renshaw, AA, Gordon, M, Corless, CL. Immunohistochemistry of unclassified sex cord-stromal tumors of the testis with a predominance of spindle cells. Mod Pathol 10, 693–700 (1997).
Berney DM, Cree I, Rao V, Moch H, Srigley JR, Tsuzuki T, et al. An Introduction to the WHO 5(th) Edition 2022 Classification of Testicular tumours. Histopathology. 2022 May. https://doi.org/10.1111/his.14675. Epub ahead of print.
Idrees, MT, Ulbright, TM, Oliva, E, Young, RH, Montironi, R, Egevad, L, et al. The World Health Organization 2016 classification of testicular non-germ cell tumours: a review and update from the International Society of Urological Pathology Testis Consultation Panel. Histopathology 70, 513-521 (2017).
Rizzo, NM, Sholl, LM, Idrees, MT, Cheville, JC, Gupta, S, Cornejo, KM, et al. Comparative molecular analysis of testicular Leydig cell tumors demonstrates distinct subsets of neoplasms with aggressive histopathologic features. Mod Pathol 34, 1935–46 (2021).
Perrone, F, Bertolotti, A, Montemurro, G, Paolini, B, Pierotti, MA, Colecchia, M. Frequent mutation and nuclear localization of β-catenin in sertoli cell tumors of the testis. Am J Surg Pathol 38, 66–71 (2014).
Tatsi, C, Faucz, FR, Blavakis, E, Carneiro, BA, Lyssikatos, C, Belyavskaya, E, et al. Somatic PRKAR1A gene mutation in a nonsyndromic metastatic large cell calcifying sertoli cell tumor. J Endocr Soc 3, 1375-1382 (2019).
Stewart, CJR, Alexiadis, M, Crook, ML, Fuller, PJ. An immunohistochemical and molecular analysis of problematic and unclassified ovarian sex cord-stromal tumors. Hum Pathol 44, 2774–81 (2013).
Lima, JF, Jin, L, De Araujo, ARC, Erikson-Johnson, MR, Oliveira, AM, Sebo, TJ, et al. FOXL2 mutations in granulosa cell tumors occurring in males. Arch Pathol Lab Med 136, 825–8 (2012).
Siegmund, S, Sholl, LM, Cornejo, KM, Sangoi, AR, Otis, CN, Mehra, R, et al. Molecular assessment of testicular adult granulosa cell tumor demonstrates significant differences when compared to ovarian counterparts. Mod Pathol 35, 697–704 (2022).
Garcia, EP, Minkovsky, A, Jia, Y, Ducar, MD, Shivdasani, P, Gong, X, et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med 141, 751–8 (2017).
Sholl, LM, Do, K, Shivdasani, P, Cerami, E, Dubuc, AM, Kuo, FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, 1–19 (2016).
Abo, RP, Ducar, M, Garcia, EP, Thorner, AR, Rojas-Rudilla, V, Lin, L. et al. BreaKmer: Detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res 43, e19, https://doi.org/10.1093/nar/gku1211 (2015).
Christakis, AG, Papke, DJ, Nowak, JA, Yurgelun, MB, Agoston, AT, Lindeman, NI, et al. Targeted cancer next-generation sequencing as a primary screening tool for microsatellite instability and lynch syndrome in upper gastrointestinal tract cancers. Cancer Epidemiol Biomark Prev 28, 1246–51 (2019).
Papke, DJ, Nowak, JA, Yurgelun, MB, Frieden, A, Srivastava, A, Lindeman, NI, et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod Pathol 31, 1882–90 (2018).
Siegmund, SE, Manning, DK, Davineni, PK, Dong, F. Deriving tumor purity from cancer next generation sequencing data: applications for quantitative ERBB2 (HER2) copy number analysis and germline inference of BRCA1 and BRCA2 mutations. Mod Pathol. 35, 1458–1467 (2022).
Serrano, J, Snuderl, M. Whole genome DNA methylation analysis of human glioblastoma using Illumina BeadArrays BT. Methods Mol Biol 1741, 31-51 (2018).
Aryee, MJ, Jaffe, AE, Corrada-Bravo, H, Ladd-Acosta, C, Feinberg, AP, Hansen, KD, et al. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–9 (2014).
Gu, Z, Eils, R, Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–9 (2016).
Polakis, P. Casein kinase 1: a Wnt’er of disconnect. Curr Biol 12, R499–501 (2002).
Provost, E, McCabe, A, Stern, J, Lizardi, I, D’Aquila, TG, Rimm, DL. Functional correlates of mutation of the Asp32 and Gly34 residues of beta-catenin. Oncogene 24, 2667–76 (2005).
Al-Obaidy, KI, Idrees, MT. Testicular tumors: a contemporary update on morphologic, immunohistochemical and molecular features. Adv Anat Pathol 28, 258–75 (2021).
Silberstein, JL, Bazzi, WM, Vertosick, E, Carver, BS, Bosl, GJ, Feldman, DR, et al. Clinical outcomes of local and metastatic testicular sex cord-stromal tumors. J Urol 192, 415–9 (2014).
Acar, C, Gurocak, S, Sozen, S. Current treatment of testicular sex cord-stromal tumors: critical review. Urology 73, 1165–71 (2009).
Hanson, JA, Ambaye, AB. Adult testicular granulosa cell tumor: A review of the literature for clinicopathologic predictors of malignancy. Arch Pathol Lab Med 135, 143–6 (2011).
Fankhauser, CD, Grogg, JB, Hayoz, S, Wettstein, MS, Dieckmann, K-P, Sulser, T, et al. Risk factors and treatment outcomes of 1375 patients with testicular leydig cell tumors: analysis of published case series data. J Urol 203, 949–56 (2020).
Young, RH, Koelliker, DD, Scully, RE. Sertoli cell tumors of the testis, not otherwise specified: a clinicopathologic analysis of 60 cases. Am J Surg Pathol 22, 709–21 (1998).
Grogg, JB, Schneider, K, Bode, PK, Kranzbühler, B, Eberli, D, Sulser, T, et al. Risk factors and treatment outcomes of 239 patients with testicular granulosa cell tumors: a systematic review of published case series data. J Cancer Res Clin Oncol 146, 2829–41 (2020).
Eble, JN, Hull, MT, Warfel, KA, Donohue, JP. Malignant sex cord-stromal tumor of testis. J Urol 131, 546–50 (1984).
Boyer, A, Paquet, M, Laguë, MN, Hermo, L, Boerboom, D. Dysregulation of WNT/CTNNB1 and PI3K/AKT signaling in testicular stromal cells causes granulosa cell tumor of the testis. Carcinogenesis 30, 869–78 (2009).
Zhang, C, Ulbright, TM. Nuclear localization of β-catenin in Sertoli cell tumors and other sex cord-stromal tumors of the testis: An immunohistochemical study of 87 cases. Am J Surg Pathol 39, 1390–4 (2015).
Colecchia, M, Bertolotti, A, Paolini, B, Giunchi, F, Necchi, A, Paganoni, AM, et al. The Leydig cell tumour Scaled Score (LeSS): a method to distinguish benign from malignant cases, with additional correlation with MDM2 and CDK4 amplification. Histopathology 78, 290-299 (2021).
Necchi, A, Bratslavsky, G, Shapiro, O, Elvin, JA, Vergilio, J-AA, Killian, JK, et al. Genomic features of metastatic testicular sex cord stromal tumors. Eur Urol Focus 5, 748–55 (2019).
Anderson, WJ, Gordetsky, JB, Idrees, MT, Al-Obaidy, KI, Kao, CS, Cornejo, KM, et al. Large cell calcifying Sertoli cell tumour: a contemporary multi-institutional case series highlighting the diagnostic utility of PRKAR1A immunohistochemistry. Histopathology 80, 677–85 (2022).
Hovestadt, V, Jones, DTW, Picelli, S, Wang, W, Kool, M, Northcott, PA, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–41 (2014).
Koelsche, C, Schrimpf, D, Stichel, D, Sill, M, Sahm, F, Reuss, DE, et al. Sarcoma classification by DNA methylation profiling. Nat Commun 12, 498 (2021).
Capper, D, Jones, DTW, Sill, M, Hovestadt, V, Schrimpf, D, Sturm, D, et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–74 (2018).
Conlon, N, Schultheis, AM, Piscuoglio, S, Silva, A, Guerra, E, Tornos, C, et al. A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod Pathol 28, 1603–12 (2015).
Yang, DT, Lowichik, A, Chen, J, Snow, BW, Ulbright, TM, Chen, Z. Cytogenetics of a pediatric unclassified sex cord-stromal tumor of the testis: a case report. Cancer Genet Cytogenet 156, 80–2 (2005).
Acknowledgements
The authors would like to acknowledge Dr. Roanh Le Dinh (Center for Research and Early Detection of Cancer, Vietnam), Dr. Igor Odintsov (Brigham & Women’s Hospital, Boston, MA), and Dr. José Manuel Lopes [Institute of Molecular Pathology and Immunology (IPATIMUP) Porto, Portugal], for their contributions to this work. They also wish to recognize the Brigham and Women’s Hospital Immunohistochemistry Laboratories (under the supervision of Ms. Mei Zheng) and the Center for Molecular Diagnostics at Brigham and Women’s Hospital. DNA methylation profiling at NYU was supported by a grant from the Friedberg Charitable Foundation (to M.S.).
Funding
Financial support for this study was provided by the Department of Pathology of Brigham and Women’s Hospital.
Author information
Authors and Affiliations
Contributions
AA designed the research study and collected tissue samples. CF, KC, MI, KA, KC, JG, SW, MSH, KT, AY, WA, GQ, CM, and SC contributed cases. SS, HT, LS, and AA performed the research and molecular analysis. YY, VV, IT, and MS performed the methylation analysis. SS and AA wrote the manuscript, and all authors provided feedback and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional Disclosures
This study was presented in part at the 2022 United States and Canadian Academy of Pathology Annual Meeting (Los Angeles, California).
Ethics approval/consent to participate
This study was performed with approval of the Institutional Review Boards of Brigham and Women’s Hospital (MGB Insight 4.0, protocol #2021P002289)
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Siegmund, S.E., Sholl, L.M., Tsai, H.K. et al. Clinicopathologic and molecular spectrum of testicular sex cord-stromal tumors not amenable to specific histopathologic subclassification. Mod Pathol 35, 1944–1954 (2022). https://doi.org/10.1038/s41379-022-01155-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01155-y
