
Abstract
Herein we evaluated a series of 21 embryonal rhabdomyosarcomas of the uterine corpus (ucERMS), a rare neoplasm, to characterize their morphology, genomics, and behavior. Patients ranged from 27 to 73 (median 52) years and tumors from 4 to 15 (median 9) cm, with extrauterine disease noted in two. Follow-up (median 16 months) was available for 14/21 patients; nine were alive and well, four died of disease, and one died from other causes. Most tumors (16/21) showed predominantly classic morphology, comprised of alternating hyper- and hypocellular areas of primitive small cells and differentiating rhabdomyoblasts in a loose myxoid/edematous stroma. A cambium layer was noted in all; seven had heterologous elements (six with fetal-type cartilage) and eight displayed focal anaplasia. The remaining five neoplasms showed only a minor component (≤20%) of classic morphology, with anaplasia noted in four and tumor cell necrosis in three. The most frequent mutations detected were in DICER1 (14/21), TP53 (7/20), PI3K/AKT/mTOR pathway (7/20), and KRAS/NRAS (5/20). Copy-number alterations were present in 10/19 tumors. Overall, 8/14 DICER1-associated ucERMS showed concurrent loss of function and hotspot mutations in DICER1, which is a feature more likely to be seen in tumors associated with DICER1 syndrome. Germline data were available for two patients, both DICER1 wild type (one with concurrent loss of function and hotspot alterations). DICER1-associated ucERMS were more likely to show a classic histological appearance including heterologous elements than DICER1-independent tumors. No differences in survival were noted between the two groups, but both patients with extrauterine disease at diagnosis and two with recurrences died from disease. As no patients had a known personal or family history of DICER1 syndrome, we favor most DICER1-associated ucERMS to be sporadic.
Similar content being viewed by others
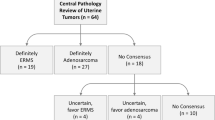

Introduction
Embryonal rhabdomyosarcoma (ERMS) in the female genital tract is most commonly seen in the cervix and vagina, typically occurring in young patients [1,2,3]. This tumor is often characterized by a “botryoid” gross appearance. Histologically it is usually composed of a proliferation of primitive undifferentiated small cells admixed with differentiating rhabdomyoblasts. Cellular areas alternate with myxomatous to edematous regions and may form a band-like/cambium layer below the surface epithelium and around entrapped, often inactive glands. Heterologous elements, primarily fetal-type cartilage, and DICER1 mutations are typical of cervical ERMS (cERMS), but not those of the vagina [4,5,6,7]. cERMS is a well-recognized manifestation of DICER1 syndrome, a hereditary condition characterized by the development of unusual neoplasms and hyperplastic lesions (including pleuropulmonary blastoma, multinodular goiter, cystic nephroma, and ovarian Sertoli–Leydig cell tumor) in young patients with germline loss-of-function variants in DICER1 [8]. In these syndromic cases, the “first hit” is the germline loss-of-function DICER1 alteration while somatic hotspot variants in the RNaseIIIb domain of DICER1 represent the “second hit” [9]. In rare instances, non-syndromic cERMS with two somatic DICER1 mutations have also been reported [4, 10]. ERMS of the uterine corpus (ucERMS) is much less common than its cervical or vaginal counterparts and information about this tumor is mostly limited to sporadic case reports or small numbers in series reporting predominantly cERMS [4, 11,12,13,14,15,16,17,18,19,20,21,22,23,24]. Herein we describe the morphology, genomics, and behavior of the largest series of ucERMS reported to date to better define its features.
Materials and methods
After approval by the institutional review board, we collected 21 ucERMS (one previously reported [21]) from the institutional archives and consult files of the authors. Only tumors originating in the uterine corpus without cervical involvement were included. Age, clinical presentation, personal/family history of DICER1-associated lesions, adjuvant therapy, follow-up, gross appearance, tumor size, and documentation of extrauterine disease at diagnosis were retrieved from the medical records or consulting pathologist when available. The number of hematoxylin and eosin-stained slides available ranged from 1 to 35 (mean and median 9). If originally performed, immunohistochemical studies for desmin, myogenin, and/or myoD1 were reported as diffuse (≥50% staining) or focal (<50%).
For tumors where sequencing had not been previously performed (n = 19), genomic DNA was isolated from macro-dissected formalin-fixed paraffin-embedded (FFPE) sections using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. Next-generation sequencing was performed using the targeted, hybrid capture 1213-gene OncoPlus panel at the University of Chicago, as previously described [25, 26] (Supplementary File 1). Somatic mutation calling was performed across all 1213 genes using a custom in-house bioinformatics pipeline as previously described [25]. Variant review was performed by two authors with specific expertise in this area (LLR and ZO) and included filters based on population variant frequencies (Exome Aggregation Consortium, http://exac.broadinstitute.org/), variant frequencies in cancer databases (COSMIC: catalogue of somatic mutations in cancer https://cancer.sanger.ac.uk/cosmic and cBioPortal https://www.cbioportal.org/), and coding effects. Somatic variant calls were inspected using Integrated Genomics Viewer (Broad Institute, MIT Harvard, Cambridge, MA).
Two tumors were previously evaluated by either Sanger sequencing of the full DICER1 gene (case 21) at McGill University or a SeqCap custom-made panel evaluating 69 genes (41 hotspots including DICER1 (E1705, D1709, G1809, E1813) and 28 full genes; Supplementary File 2) (case 20) at the University Hospital Ghent.
Statistical analysis was conducted using GraphPad Prism (Version 8.4.3, GraphPad Software; San Diego, CA). Univariate analysis of categorical and continuous variables was performed using Fisher exact and Mann–Whitney tests, respectively. Survival was assessed by the log-rank test. All hypothesis tests were two-sided and statistical significance was set at p < 0.05.
Results
Clinicopathological features
Clinical and pathological features are summarized in Tables 1 and 2. Patients ranged from 27 to 73 (mean and median 52) years and most (16/19; unknown in two) presented with abnormal uterine bleeding (n = 9) or postmenopausal bleeding (n = 7). No patient reported a personal (n = 17) or family (n = 14) history of DICER1-associated lesions. Initial surgery was hysterectomy in 16/21 and polypectomy in five, followed by hysterectomy in the latter five patients with residual disease noted in two. All tumors were centered in the uterine corpus with five minimally extending into the lower uterine segment. They ranged from 4 to 15 (mean and median 9; size not available in the three entirely removed by curettage) cm. They varied in color, tan, white, pink, red, brown, and 11 of 15 with details were described as polypoid or botryoid (Fig. 1A). Hemorrhage was observed in four and necrosis in three. Extrauterine disease was present in 2/21 patients. It was localized in one (left adnexa), while the other had metastases to the omentum and other abdominopelvic sites and underwent subtotal debulking. The 2015 International Federation of Gynecology and Obstetrics (FIGO) stage (uterine sarcoma staging) was as follows: I (3/21; cannot be further subclassified as size not available), IA (4/21), IB (12/21), IIA (1/21), and IIIB (1/21).

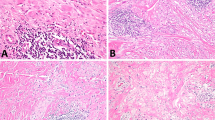
Polypoid to grape-like fleshy mass with extensive hemorrhage and necrosis (A, left macroscopic, right microscopic). Variably sized aggregates of hyperchromatic cells are present in a myxoid stroma (B). Heterologous fetal-type cartilage (C). Primitive cells surrounding an inactive endometrial gland vaguely mimics the appearance of adenosarcoma (D).
Tumors from hysterectomy specimens showed a destructive pattern of invasion into the myometrium that was limited (<1 mm) in 4/18, <50% in 11/18, >50% in 2/18, or transmural with serosal involvement in one. Most tumors (16/21) were comprised of alternating hypocellular and hypercellular areas (Figs. 1B–D and 2). In these neoplasms, the hypocellular foci typically displayed myxoid/edematous stroma, but occasionally collagenous (ranging from loose to dense) stroma was noted. Five tumors (31%) were primarily hypocellular. One tumor with exuberant botryoid growth was hypocellular in the superficial (polypoid) areas, but hypercellular in the remainder of the neoplasm. Hypercellular areas contained a haphazard distribution of variably sized cellular aggregates of small cells often forming a subepithelial band (cambium layer) and surrounding scattered entrapped small inactive and sometimes dilated endometrial glands (n = 15).

Primitive cells with brisk mitoses and apoptotic activity beneath the surface epithelium (A), differentiating rhabdomyoblasts with discernible cross-striations (arrow) and tadpole cells (right) (B), spider cells (C), and anaplasia (D).
Tumors displayed an admixture of primitive “blue” cells and differentiating rhabdomyoblasts. The primitive cells typically predominated (n = 15) and were small, ovoid, or spindled with scant cytoplasm, hyperchromatic nuclei, and variably prominent nucleoli. Differentiating rhabdomyoblasts often showed abundant eosinophilic cytoplasm, eccentric round nuclei, and prominent nucleoli, but occasionally had elongated cytoplasmic tails (strap or “tadpole” cells). In two tumors, polygonal cells with finely granular eosinophilic to vacuolated cytoplasm and peripherally displaced ovoid nuclei (“spider cells”) (10% and 50% of the neoplasm, respectively), reminiscent of adult-type rhabdomyoma were noted. Anaplasia, characterized by markedly enlarged, atypical cells with hyperchromatic nuclei and bizarre multipolar mitoses [27], was observed in 8/16, but accounted for ≤25% of the tumor. Mitoses ranged from 1 to 65 (mean and median 18) per 10 high-power fields and brisk apoptotic activity was noted in 15/16. Mitoses and apoptosis were most conspicuous among primitive cells. Seven of these tumors contained one or more heterologous elements: fetal-type cartilage (n = 6), mature adipose tissue (n = 2), neuroectodermal rosettes (n = 1), and/or mature osteoid (n = 1), the latter three as microscopic foci representing <1% of the neoplasm. Cartilaginous nodules were typically small, rounded (~1 mm), and only found on a few slides (range 1–3), but one tumor showed confluent foci (up to 9 mm) present on 20 out of 35 slides. One tumor showed minor areas (<5%) resembling alveolar rhabdomyosarcoma. Overall, vasculature consisted of small to medium, thin-to-thick-walled vessels that were occasionally dilated. Three tumors had large, thrombosed thick-walled vessels while two others focally showed a “chicken-wire” appearance. Infarct-type necrosis was present in 10/16 tumors, but tumor cell necrosis was not identified. Lymphovascular invasion was noted in one tumor associated with extrauterine disease.
The remaining five tumors (Fig. 3) displayed only focal (≤20%) alternating hyper- and hypocellular areas, and one had a hint of a cambium layer. Four were composed of sheets (admixed with fascicles in one) of primitive cells and differentiating rhabdomyoblasts with minimal intervening stroma, while the fifth (case 14) had a fascicular growth of spindle cells with varying cellularity set in a strikingly myxoid matrix. Scattered myxoid pools as well as rare foci (<5%) resembling alveolar rhabdomyosarcoma were present in one tumor, but none contained heterologous elements. Anaplasia was noted in four tumors (ranging from 5 to 25%), mitoses ranged from 8 to 26 (mean 17, median 18) per 10 high-power fields, and all showed brisk apoptotic activity. Tumor cell necrosis was present in three and lymphovascular invasion in two tumors.

Barely discernible rounded aggregates in an overall hypercellular tumor (A). Sheets of primitive cells with large eosinophilic multinucleated giant cells (B). Variably cellular areas with a myxoedematous background (C) and vague storiform appearance (D).
Twelve ucERMS for which stains were available were positive for desmin (10 diffuse, 2 focal). Myogenin and myoD1 were often focally positive (8/13 and 7/8, respectively), with the remaining tumors showing diffuse expression.
Other pathologic findings included leiomyomas (n = 6), atypical hyperplasia (n = 2), adenomyosis (n = 2), and endometrial polyp, adenomatoid tumor, and cervical mesonephric hyperplasia (each n = 1).
Molecular features
DICER1 mutations were detected in 14/21 ucERMS (Fig. 4A and Supplementary File 3), with DICER1 sequencing depth averaging 501 (range 192–921). Germline testing had been previously performed in two patients (cases 1 and 2), and both were DICER1 wild type. Among DICER1-associated ucERMS, 8/14 showed concurrent loss of function and hotspot alterations (including case 2), while 6/14 had hotspot alterations only (including case 1). TP53 mutations were detected in 7/20 tumors, PI3K/AKT/mTOR pathway mutations including KMT2D, KMT2C, PPP2R1A, PIK3CA, PIK3R1, ARID1A, ARID4A, MTOR, and PTEN in 7/20, and KRAS/NRAS mutations in 5/20. For DICER1-associated ucERMS, additional mutations included those in the PI3K/AKT/mTOR pathway (4/13), KRAS (4/13), TP53 (3/13), and EP300 (1/12). DICER1-independent ucERMS showed TP53 (4/7), PI3K/AKT/mTOR pathway (3/7), BCOR (1/6), NRAS (1/7), FBXW7 (1/7), and HIST1H3B (1/7) mutations. The ucERMS sequenced on the SeqCap-targeted panel (case 20) did not harbor any mutations.

Pathogenic mutations (A) and copy-number alterations (B).
Copy-number alterations were detected in 10/19 tumors including both DICER1-associated (5/11) and DICER1-independent (5/11) (Fig. 4B). Among these, all but three (two DICER1-associated and one DICER1-independent) had accompanying TP53 mutations. The most common copy-number alterations were genomic losses involving FGFR3 (4p16.3), GNA11 and STK11 (19p13.3), and TSC2 (16p13.3). Copy-number gains in RAD21 (8q24.11) and MYC (8q24.21) were detected in one tumor (case 5). No DICER1 copy-number alterations were identified. As cases 20 and 21 were not sequenced by the Oncoplus panel, copy-number alterations were not assessed.
Follow-up
Follow-up of at least 6 months was available for 14/21 patients, ranging from 6 to 97 (mean 29, median 16) months, being >1 year for 10 patients. Eight patients received adjuvant chemotherapy, in one accompanied by whole pelvic radiation and a vaginal boost, while three did not undergo further treatment (one polyp-confined, two with <50% invasion). No details regarding adjuvant therapy were available for the other three patients. Recurrences were noted in 2/14 patients (omentum or peritoneum/sigmoid colon, both FIGO stage IB at presentation), while the patient with extensive metastases at diagnosis who underwent incomplete resection (FIGO stage IIIB, case 2) showed disease progression. These three patients and the other with extrauterine disease at diagnosis (FIGO stage IIA, case 9) died of disease 6–21 months after original diagnosis while the remaining patients (all FIGO stage I) were alive without evidence of disease (n = 9) or died from other causes (n = 1).
Statistical analysis
As the entire coding sequence of DICER1 was not sequenced in one tumor (case 20), it was excluded from the DICER1 statistical analysis. DICER1-associated ucERMS were more likely to show a classic histological appearance (p < 0.01), cambium layer (p < 0.01), and heterologous elements (p = 0.05). Younger age also trended toward statistical significance in this cohort (p = 0.07). Tumor size, anaplasia, mitotic index, TP53 status, presence of extrauterine disease/recurrence, and outcome did not differ between the two groups (Table 3). Both patients with extrauterine disease at diagnosis, as well as the two that recurred died from disease (p < 0.01), but no other clinicopathological features were predictive of survival.
Discussion
Pure rhabdomyosarcomas of the uterine corpus are uncommon being first reported in the late 19th century based on an early literature review by Robertson [28] who himself described a rhabdomyosarcoma based in the lower uterine segment/upper endocervix. A table in that report summarizes 13 previously reported uterine (corpus and cervix) rhabdomyosarcomas and documents three uterine corpus tumors with heterologous elements (cartilage and/or fat), two of which also showed myxomatous stroma. As the subclassification system for rhabdomyosarcomas was non-existent at that time, the presence of heterologous elements allows one to infer they likely represented ucERMS assuming they were well sampled. Aside from these cases, a review of the literature resulted in identification of 23 ucERMS with available morphological or molecular findings (Supplementary File 4) [4, 11,12,13,14,15,16,17,18,19,20,21,22,23,24]. However, as morphological descriptions were limited in most, and molecular testing was only performed in four, relatively little is known about their pathogenesis, morphologic features, and behavior in contrast to their cervical counterparts. In this study, our analysis of 21 ucERMS provides a detailed morphological analysis and a comprehensive review of their molecular profile with evidence of a morphologic-molecular correlation.
ERMS are most common in the head and neck and genitourinary tract, but occasionally arise in the biliary tract, retroperitoneum, abdomen, pelvis, perineum, and visceral organs [29]. Morphologically, most (76% herein) ucERMS resemble ERMS in other locations, consisting of primitive cells and differentiating rhabdomyoblasts in a loose myxoedematous stroma with alternating areas of hyper- and hypocellularity [30]. As the botryoid variant of ERMS arises beneath an epithelial-lined surface, it is not surprising that many ucERMS showed the polypoid growth and cambium layer [30] characteristic of this variant akin to their counterparts in the vagina, cervix, bladder, prostate, biliary tract, and head and neck [29]. Heterologous elements, in particular, fetal-type cartilage, have been reported in ~45% of cERMS [1, 4, 22], but this is an infrequent finding in extrauterine ERMS with only rare examples reported in the ovary, peritoneum, fallopian tube, bladder, and mandible [31,32,33,34,35,36,37,38]. Previously, heterologous elements were described in 29% of ucERMS (Supplementary File 4), which mirrors the 33% observed in this study. Anaplasia has been reported in 48/259 childhood ERMS (including botryoid variant) [39], but was noted in over half (57%) of our ucERMS. The explanation behind this nearly threefold difference is unclear but may possibly be related to the number of slides examined (not provided in the other study), the focality of this finding, not been considered in other studies, or it could be a site-specific phenomenon. Two ucERMS had areas resembling adult-type rhabdomyoma, a feature previously termed highly differentiated rhabdomyosarcoma when diffuse [40], which in one study of 24 pediatric rhabdomyosarcomas showed a 100% overall survival rate with a median follow-up of 4.6 years [41]. Similarly, one patient with this finding (which comprised ~50% of the tumor) in the current series is alive and well 57 months after diagnosis; no follow-up was available for the other patient in which it represented ~10% of the tumor as the case is recent.
The focus on ERMS of the uterus has been in tumors occurring in the cervix as they are by far more common than those of the corpus. Although cERMS is most frequent in the pediatric population, it may occur in reproductive age and postmenopausal women [22], similar to that seen for ucERMS. The gross and microscopic appearance closely overlaps, and careful examination is necessary to deduce the site of origin. It is difficult to compare the prognosis of ucERMS to cERMS as studies focusing specifically on the latter are sparse. Nonetheless, in the two largest series to date, comprised of 13 and 14 cERMS, respectively, no patients had extrauterine disease at presentation and only one died from her disease [1, 2].
As in the cervix, the main and most challenging differential diagnosis of the ucERMS is with Mullerian adenosarcoma. ucERMS is typically misdiagnosed as adenosarcoma, as the latter is much more common in the corpus and both tumors share a polypoid growth, condensation of malignant stroma around the epithelium, and fetal-type cartilage [42]. Further confounding the differential is that a subset of adenosarcomas may show rhabdomyoblastic (embryonal and other subtypes) differentiation and in some it may be extensive [4, 43,44,45,46,47,48,49]. In the largest study of adenosarcomas to date, 15/22 with heterologous elements were comprised of an extensive component of ERMS, with 10/15 showing additional heterologous elements such as fetal cartilage and/or adipose tissue [42]. However, adenosarcoma is defined as a biphasic tumor in which glands are part of the neoplasm, and often have a phyllodes-like architecture with intraluminal polypoid projections. The glands frequently show hyperplastic or metaplastic changes and the low-grade sarcoma typically has a fibroblastic or endometrial stromal morphology with a low mitotic index and absence of apoptotic cells [42, 43]. In contrast, in ERMS, the cells in the cambium layer and around pre-existing entrapped glands are primitive, associated with brisk mitoses and abundant apoptotic bodies. Some high-grade adenosarcomas have been reported but in these cases the stromal cells are pleomorphic and not primitive [48]. Recently, several series have detected DICER1 alterations in presumed adenosarcomas, including 8/20 with rhabdomyoblastic differentiation (Supplementary File 5) [4, 46,47,48,49,50]. Based on the provided images from two of these studies [48, 49], at least some of them we would favor to likely represent pure ucERMS with entrapped glands. Furthermore, none of the amplifications typical of adenosarcoma, including MDM2/CDK4/HMGA2, TERT, or MYBL1, have been detected in ERMS [46, 47].
ucERMS with neuroectodermal differentiation may pose a different diagnostic challenge. Although such differentiation is uncommon, it has been reported in adenosarcoma and carcinosarcoma, as well as in ERMS [51]. To establish a diagnosis of adenosarcoma or carcinosarcoma, the typical diagnostic features should be present. In our series, the tumors were extensively sampled, and these features were not identified. If the neuroectodermal component was extensive, a peripheral neuroectodermal tumor [52] could enter in the differential diagnosis.
As five ucERMS lacked a dominant hypercellular–hypocellular growth and most did not have a cambium layer, whether these are ERMS or another rhabdomyosarcoma subtype merits discussion. An alveolar appearance was focally identified (<5%) in one variant ucERMS but also in a classic ucERMS (also <5%). It is well-recognized that rhabdomyosarcomas may show mixed embryonal and alveolar features [7]; however, our cases were extensively sampled and the alveolar morphology was minimal. While our panel did not test for the characteristic PAX3-FOXO1 or PAX7-FOXO1 fusions, we were able to exclude this rhabdomyosarcoma subtype based on morphology. Furthermore, alveolar rhabdomyosarcoma is extremely uncommon in the uterine corpus, with examples limited to case reports or small series comprised of different rhabdomyosarcoma subtypes [53,54,55,56]. Features suggestive of spindle cell/sclerosing rhabdomyosarcoma including a leiomyosarcoma/fibrosarcoma-like appearance, prominent hyalinization/sclerosis, or a pseudovascular growth pattern were not observed either [57]. While our panel did not test for the characteristic MYOD1 mutation or NCOA2/VGLL2 rearrangement, based on morphology we were able to exclude this rhabdomyosarcoma subtype. This subtype has also received limited attention in the gynecologic tract [17, 58] and is currently not recognized in the 2020 WHO Female Genital Tumors [59]. There are no defining morphologic or molecular features for the pleomorphic subtype of rhabdomyosarcoma other than the presence of markedly pleomorphic cells [60, 61]. While anaplasia was present in 80% of these five ucERMS, all had a component of primitive cells comprising >50% of the tumor, which in our opinion, excludes the diagnosis of pleomorphic rhabdomyosarcoma. Other diagnostic considerations for these tumors could include carcinosarcoma or adenosarcoma with extensive rhabdomyoblastic differentiation or with sarcomatous overgrowth. As mentioned previously, these tumors were extensively sampled and the typical diagnostic features of carcinosarcoma or adenosarcoma were not identified.
DICER1 is a ribonuclease (RNase) III endoribonuclease that cleaves precursor microRNAs into mature microRNAs, which regulate gene expression [8]. Germline DICER1 mutations were first identified in pediatric patients with familial pleuropulmonary blastoma [62], but as the disease spectrum markedly expanded, it was renamed to DICER1 syndrome. In the gynecological tract, the most common manifestations include cERMS and Sertoli–Leydig cell tumor, which occur most often in adolescence or early adulthood in contrast to most other lesions that have a younger onset [8]. Aside from cERMS, the presence of fetal-type cartilage has also been reported in other lesions associated with DICER1 syndrome including Sertoli–Leydig cell tumor, pleuropulmonary blastoma, anaplastic sarcoma of the kidney, Wilms tumor, nasal chondromesenchymal hamartoma, DICER1-associated central nervous system sarcoma, and pleuropulmonary blastoma-like peritoneal sarcoma [63,64,65,66,67,68]. In mice, miRNAs and Dicer play important roles in chondrogenesis [69,70,71], suggesting a connection with the presence of cartilage in DICER1-associated tumors. Exactly how DICER1 mutations contribute to cartilaginous differentiation in these tumors is currently unknown. Given their marked morphological overlap with cERMS, it is not surprising that 88% of our classic-appearing ucERMS harbored a DICER1 mutation. In one of the two lacking a DICER1 mutation, only the hotspot regions were evaluated; thus, a mutation could not be excluded. Heterologous elements were only identified in DICER1-associated ucERMS and were present at a similar frequency (50%) as seen in cERMS.
Anaplasia was noted in both DICER1-associated and DICER1-independent ucERMS, including all those harboring concurrent TP53 mutations. An association between anaplasia and TP53 mutations has previously been described in rhabdomyosarcomas (predominantly of embryonal type) from patients with TP53 germline mutations (i.e., Li-Fraumeni syndrome) [72] as well as in other anaplastic embryonal tumors including Wilms tumor and medulloblastoma [73, 74]. However, four ucERMS with anaplasia lacked TP53 mutations, and in a fifth, TP53 was not evaluated. Interestingly, these were all DICER1-associated ucERMS where anaplasia involved <5% of the tumor. Further review of the slides matching blocks used for sequencing revealed that the anaplastic cells comprised <1% in two tumors and was absent in the other two, which may explain the lack of a TP53 mutation. Of note, one DICER1-associated ucERMS with anaplasia lacked anaplastic cells on the slide used for sequencing, but a TP53 mutation was still detected, similar to what has been described in a subset of anaplastic Wilms tumors [75]. In this study, the presence of anaplasia did not impact survival, but the literature shows mixed results in ERMS from other sites. Kodet et al. evaluated all tumors from intergroup rhabdomyosarcoma studies 1–3 (~3000 tumors) and identified 110 with anaplastic/pleomorphic features [76]. They noted no significant survival difference between tumors without anaplasia versus those with focal anaplasia (5-year survival 68% versus 60%, respectively, p = 0.177), but those with diffuse anaplasia had significantly decreased survival (5-year survival 68% versus 45%, respectively, p = 0.004). Qualman et al. found on univariate analysis an association between anaplasia (focal or diffuse) and failure-free survival as well as overall survival, but on multivariate analysis, anaplasia was not an independent predictor [39].
Outside of the gynecological tract, DICER1 mutations are exceedingly rare in ERMS, and a recurring mutation or fusion has not been identified for this group of tumors. Instead, these sarcomas are genomically diverse with previously reported alterations in the PI3K pathway, RAS pathway, BCOR, CTNNB1, and FBXW7, as well as aneuploidy [7, 77,78,79]. Similarly, in our study, 35% and 25% (not tested in one) of ucERMS, including both DICER1-associated and DICER1-independent tumors, harbored mutations involving the PI3K/AKT/mTOR or RAS pathway, respectively. One DICER1-independent ucERMS with an NRAS mutation (case 3) also harbored a FBXW7 mutation. Another DICER1-independent ucERMS (case 14) had a BCOR frameshift mutation, but aside from a copy-number loss in CHEK2, did not show other genomic abnormalities. This tumor was characterized by prominent myxoid stroma, abundant spindled cells, and infrequent differentiating rhabdomyoblasts, features which in aggregate were not present in other ucERMS. While the morphology of BCOR-mutant ERMS outside of the gynecologic tract has not been described to our knowledge, within the uterine corpus, this histology might raise the possibility of BCOR-related high-grade endometrial stromal sarcoma. However, rhabdomyoblastic differentiation and positivity for skeletal muscle markers are not features of high-grade endometrial stromal sarcomas, and only BCOR fusions or internal tandem duplication have been described in these tumors [80, 81], in contrast to the frameshift mutation present in this case.
As many of our tumors occurred relatively recently, follow-up was limited and a detailed correlation between clinicopathological features and outcome was not feasible. We did note that both tumors with extrauterine disease at diagnosis and two others with recurrences resulted in death. In contrast, there were no disease-related deaths in any other patients (all stage I at diagnosis). Similarly, in previously reported ucERMS with follow-up data, 3/15 patients died from disease and all had metastases at diagnosis (n = 1) or recurrences (n = 2) [18,19,20]. Notably, progression-free survival was extremely short in the four patients herein with extrauterine disease (4–5 months), highlighting the aggressive nature of this tumor once outside the uterus.
One limitation of our study is that germline data were only available in two patients, neither of whom harbored a germline DICER1 mutation. The mean and median age of our cohort was 52 years, which is significantly older than most patients with DICER1 syndrome-related cERMS. However, five patients were ≤40 years, and four had tumors that harbored a DICER1 mutation. Two had no personal or family history of DICER1-related lesions, but no information was available for the others. An indirect way to “screen” for syndromic cases based on sequencing data of the tumor genome, would be to look at cases with concurrent loss of function (presumed germline) and hotspot (presumed somatic) variants, because frequent somatic (tumor-only) hotspot variants in DICER1 have been described as a “second hit” to the germline loss-of-function DICER1 alteration in DICER1 syndromic cases [9]. In our series, 8/14 tumors had such concurrent mutations and only one of them had germline testing, but both alterations were found to be somatic. The remaining patient with germline testing only had a hotspot mutation, as would be expected in sporadic tumors. To our knowledge, the only previously reported ucERMS likely associated with a germline DICER1 mutation (as she also had a cystic nephroma) occurred in a 10-year-old girl whose tumor was not sequenced [24]. Thus, we suspect that the vast majority of ucERMS with DICER1 mutations are likely somatic in origin, but it is imperative to always be aware of the potential association with DICER1 syndrome, especially in younger patients or those with a personal or family history of other DICER1-associated neoplasms. Nonetheless, in ~10–20% of patients with DICER1 syndrome, the germline DICER1 mutation can arise de novo [82]. Due to the rarity of these tumors and unavailability of routine DICER1 sequencing as an intermediary screening step, the safest approach would be formal referral of all patients for genetic counseling as is recommended for patients with ovarian Sertoli–Leydig cell tumors [83, 84].
In summary, we identified somatic DICER1 mutations in 67% of ucERMS. Tumors with DICER1 mutations are more likely to have a classic morphological appearance, cambium layer, and heterologous elements than DICER1-independent ucERMS. While follow-up was limited, we did not detect any survival differences between the two groups; however, patients with extrauterine disease at diagnosis or recurrences died from disease. Although we favor most DICER1-associated ucERMS to be sporadic, identification of one should always prompt further evaluation to exclude DICER1 syndrome.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Daya DA, Scully RE. Sarcoma botryoides of the uterine cervix in young women: a clinicopathological study of 13 cases. Gynecol Oncol. 1988;29:290–304.
Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol. 2012;25:602–14.
Kirsch CH, Goodman M, Esiashvili N. Outcome of female pediatric patients diagnosed with genital tract rhabdomyosarcoma based on analysis of cases registered in SEER database between 1973 and 2006. Am J Clin Oncol. 2014;37:47–50.
de Kock L, Yoon JY, Apellaniz-Ruiz M, Pelletier D, McCluggage WG, Stewart CJR, et al. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol. 2020. https://doi.org/10.1038/s41379-019-0436-0.
Leuschner I, Harms D, Mattke A, Koscielniak E, Treuner J. Rhabdomyosarcoma of the urinary bladder and vagina: a clinicopathologic study with emphasis on recurrent disease: a report from the Kiel Pediatric Tumor Registry and the German CWS Study. Am J Surg Pathol. 2001;25:856–64.
Foulkes WD, Bahubeshi A, Hamel N, Pasini B, Asioli S, Baynam G, et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat. 2011;32:1381–4.
Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–31.
Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–72.
de Kock L, Wu MK, Foulkes WD. Ten years of DICER1 mutations: provenance, distribution, and associated phenotypes. Hum Mutat. 2019;40:1939–53.
de Kock L, Boshari T, Martinelli F, Wojcik E, Niedziela M, Foulkes WD. Adult-onset cervical embryonal rhabdomyosarcoma and DICER1 mutations. J Low Genit Trac Dis. 2016;20:e8–10.
Donkers B, Kazzaz BA, Meijering JH. Rhabdomyosarcoma of the corpus uteri. Report of two cases with review of the literature. Am J Obstet Gynecol. 1972;114:1025–30.
Hart WR, Craig JR. Rhabdomyosarcomas of the uterus. Am J Clin Pathol. 1978;70:217–23.
Montag TW, D’Ablaing G, Schlaerth JB, Gaddis O Jr., Morrow CP. Embryonal rhabdomyosarcoma of the uterine corpus and cervix. Gynecol Oncol. 1986;25:171–94.
Teshima H, Narahara H, Nishida J, Takada S, Cheng SZ, Mizutani K, et al. Pure rhabdomyosarcoma of the corpus uteri: a case report with a review of the literature. Asia Ocean J Obstet Gynaecol. 1988;14:301–5.
Takano M, Kikuchi Y, Aida S, Sato K, Nagata I. Embryonal rhabdomyosarcoma of the uterine corpus in a 76-year-old patient. Gynecol Oncol. 1999;75:490–4.
Scheidt P, Moerman PH, Vergote I. Rhabdomyosarcoma of the corpus of the uterus: a case report. Eur J Gynaecol Oncol. 2000;21:371–3.
McCluggage WG, Lioe TF, McClelland HR, Lamki H. Rhabdomyosarcoma of the uterus: report of two cases, including one of the spindle cell variant. Int J Gynecol Cancer. 2002;12:128–32.
Reynolds EA, Logani S, Moller K, Horowitz IR. Embryonal rhabdomyosarcoma of the uterus in a postmenopausal woman. Case report and review of the literature. Gynecol Oncol. 2006;103:736–9.
Ferguson SE, Gerald W, Barakat RR, Chi DS, Soslow RA. Clinicopathologic features of rhabdomyosarcoma of gynecologic origin in adults. Am J Surg Pathol. 2007;31:382–9.
da Silva BB, Dos Santos AR, Bosco Parentes-Vieira J, Lopes-Costa PV, Pires CG. Embryonal rhabdomyosarcoma of the uterus associated with uterine inversion in an adolescent: a case report and published work review. J Obstet Gynaecol Res. 2008;34:735–8.
Garrett LA, Harmon DC, Schorge JO. Embryonal rhabdomyosarcoma of the uterine corpus. J Clin Oncol. 2013;31:e48–50.
Li RF, Gupta M, McCluggage WG, Ronnett BM. Embryonal rhabdomyosarcoma (botryoid type) of the uterine corpus and cervix in adult women: report of a case series and review of the literature. Am J Surg Pathol. 2013;37:344–55.
Yamada S, Harada Y, Noguchi H, Satoh N, Kimura S, Nakayama T, et al. Embryonal rhabdomyosarcoma arising from the uterine corpus in a postmenopausal female: a surgical case challenging the genuine diagnosis on a cytology specimen. Diagn Pathol. 2016;11:3.
Dural O, Kebudi R, Yavuz E, Yilmaz I, Buyukkapu Bay S, Schultz KAP, et al. DICER1-related embryonal rhabdomyosarcoma of the uterine corpus in a prepubertal girl. J Pediatr Adolesc Gynecol. 2020;33:173–6.
Kadri S, Long BC, Mujacic I, Zhen CJ, Wurst MN, Sharma S, et al. Clinical validation of a next-generation sequencing genomic oncology panel via cross-platform benchmarking against established amplicon sequencing assays. J Mol Diagn. 2017;19:43–56.
Bennett JA, Ritterhouse LL, Furtado LV, Lastra RR, Pesci A, Newell JM, et al. Female adnexal tumors of probable Wolffian origin: morphological, immunohistochemical, and molecular analysis of 15 cases. Mod Pathol. 2020;33:734–47.
Fletcher C, Bridge J, Hogendoorn P, Mertens F. WHO classification of tumours of soft tissue and bone. Lyon: World Health Organization; 2013.
Robertson AR. Rhabdomyosarcoma of the uterus; with the report of a case. J Med Res. 1909;20:297–310.
Agaram NP. Update on myogenic sarcomas. Surg Pathol Clin. 2019;12:51–62.
World Health Organization. WHO classification of tumours soft tissue and bone. Lyon: World Health Organization; 2020.
Nielsen GP, Oliva E, Young RH, Rosenberg AE, Prat J, Scully RE. Primary ovarian rhabdomyosarcoma: a report of 13 cases. Int J Gynecol Pathol. 1998;17:113–9.
Al-Shedoukhy A, Qayyum A. Urinary bladder botryoid rhabdomyosarcoma with immature cartilage in a 24-year-old male patient: a case report. Saudi J Kidney Dis Transpl. 2003;14:522–5.
Allende DS, Yang B. Primary ovarian rhabdomyosarcoma with heterologous elements: a case report. Int J Gynecol Pathol. 2008;27:402–6.
Paner GP, McKenney JK, Epstein JI, Amin MB. Rhabdomyosarcoma of the urinary bladder in adults: predilection for alveolar morphology with anaplasia and significant morphologic overlap with small cell carcinoma. Am J Surg Pathol. 2008;32:1022–8.
de Kock L, Druker H, Weber E, Hamel N, Traubici J, Malkin D, et al. Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum Pathol. 2015;46:917–22.
Peters SM, Kunkle T, Perrino MA, Philipone EM, Yoon AJ. Mandibular embryonal rhabdomyosarcoma with cartilaginous metaplasia: report of a case and review of literature. Oral Surg Oral Med Oral Pathol Oral Radio. 2017;124:e288–93.
Warren M, Hiemenz MC, Schmidt R, Shows J, Cotter J, Toll S, et al. Expanding the spectrum of dicer1-associated sarcomas. Mod Pathol. 2020;33:164–74.
McCluggage WG, Apellaniz-Ruiz M, Chong AL, Hanley KZ, Velazquez Vega JE, McVeigh TP, et al. Embryonal rhabdomyosarcoma of the ovary and fallopian tube: rare neoplasms associated with germline and somatic DICER1 mutations. Am J Surg Pathol. 2020;44:738–47.
Qualman S, Lynch J, Bridge J, Parham D, Teot L, Meyer W, et al. Prevalence and clinical impact of anaplasia in childhood rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer. 2008;113:3242–7.
Kodet R, Newton WA Jr., Hamoudi AB, Asmar L, Wharam MD, Maurer HM. Orbital rhabdomyosarcomas and related tumors in childhood: relationship of morphology to prognosis-an Intergroup Rhabdomyosarcoma study. Med Pediatr Oncol. 1997;29:51–60.
Teot LA, Schneider M, Thorner AR, Tian J, Chi YY, Ducar M, et al. Clinical and mutational spectrum of highly differentiated, paired box 3:forkhead box protein o1 fusion-negative rhabdomyosarcoma: a report from the Children’s Oncology Group. Cancer. 2018;124:1973–81.
Clement PB, Scully RE. Mullerian adenosarcoma of the uterus: a clinicopathologic analysis of 100 cases with a review of the literature. Hum Pathol. 1990;21:363–81.
Clement PB, Scully RE. Mullerian adenosarcoma of the uterus. A clinicopathologic analysis of ten cases of a distinctive type of mullerian mixed tumor. Cancer. 1974;34:1138–49.
Clement PB, Scully RE. Müllerian adenosarcomas of the uterus with sex cord-like elements. A clinicopathologic analysis of eight cases. Am J Clin Pathol. 1989;91:664–72.
Gallardo A, Prat J. Mullerian adenosarcoma: a clinicopathologic and immunohistochemical study of 55 cases challenging the existence of adenofibroma. Am J Surg Pathol. 2009;33:278–88.
Howitt BE, Sholl LM, Dal Cin P, Jia Y, Yuan L, MacConaill L, et al. Targeted genomic analysis of Mullerian adenosarcoma. J Pathol. 2015;235:37–49.
Piscuoglio S, Burke KA, Ng CK, Papanastasiou AD, Geyer FC, Macedo GS, et al. Uterine adenosarcomas are mesenchymal neoplasms. J Pathol. 2016;238:381–8.
Hodgson A, Amemiya Y, Seth A, Djordjevic B, Parra-Herran C. High-grade mullerian adenosarcoma: genomic and clinicopathologic characterization of a distinct neoplasm with prevalent TP53 pathway alterations and aggressive behavior. Am J Surg Pathol. 2017;41:1513–22.
Bean GR, Anderson J, Sangoi AR, Krings G, Garg K. DICER1 mutations are frequent in mullerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol. 2019;32:280–9.
Mullen MM, Divine LM, Hagemann IS, Babb S, Powell MA. Endometrial adenosarcoma in the setting of a germline DICER1 mutation: a case report. Gynecol Oncol Rep. 2017;20:121–4.
Cate F, Bridge JA, Crispens MA, Keedy VL, Troutman A, Coffin CM, et al. Composite uterine neoplasm with embryonal rhabdomyosarcoma and primitive neuroectodermal tumor components: rhabdomyosarcoma with divergent differentiation, variant of primitive neuroectodermal tumor, or unique entity? Hum Pathol. 2013;44:656–63.
Chiang S, Snuderl M, Kojiro-Sanada S, Quer Pi-Sunyer A, Daya D, Hayashi T, et al. Primitive neuroectodermal tumors of the female genital tract: a morphologic, immunohistochemical, and molecular study of 19 cases. Am J Surg Pathol. 2017;41:761–72.
Chiarle R, Godio L, Fusi D, Soldati T, Palestro G. Pure alveolar rhabdomyosarcoma of the corpus uteri: description of a case with increased serum level of CA-125. Gynecol Oncol. 1997;66:320–3.
Case AS, Kirby TO, Conner MG, Huh WK. A case report of rhabdomyosarcoma of the uterus associated with uterine inversion. Gynecol Oncol. 2005;96:850–3.
Fukunaga M. Pure alveolar rhabdomyosarcoma of the uterine corpus. Pathol Int. 2011;61:377–81.
Pinto A, Kahn RM, Rosenberg AE, Slomovitz B, Quick CM, Whisman MK, et al. Uterine rhabdomyosarcoma in adults. Hum Pathol. 2018;74:122–8.
Agaram NP, LaQuaglia MP, Alaggio R, Zhang L, Fujisawa Y, Ladanyi M, et al. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol. 2019;32:27–36.
Kim DW, Shin JH, Lee HJ, Hong YO, Joo JE, Kim EK. Spindle cell rhabdomyosacoma of uterus: a case study. Korean J Pathol. 2013;47:388–91.
World Health Organization. WHO classification of tumours female genital tumors. Lyon: World Health Organization; 2020.
Fadare O, Bonvicino A, Martel M, Renshaw IL, Azodi M, Parkash V. Pleomorphic rhabdomyosarcoma of the uterine corpus: a clinicopathologic study of 4 cases and a review of the literature. Int J Gynecol Pathol. 2010;29:122–34.
Ordi J, Stamatakos MD, Tavassoli FA. Pure pleomorphic rhabdomyosarcomas of the uterus. Int J Gynecol Pathol. 1997;16:369–77.
Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965.
Prat J, Young RH, Scully RE. Ovarian Sertoli–Leydig cell tumors with heterologous elements. II. Cartilage and skeletal muscle: a clinicopathologic analysis of twelve cases. Cancer. 1982;50:2465–75.
Priest JR, McDermott MB, Bhatia S, Watterson J, Manivel JC, Dehner LP. Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer. 1997;80:147–61.
Doros LA, Rossi CT, Yang J, Field A, Williams GM, Messinger Y, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol. 2014;27:1267–80.
Stewart DR, Messinger Y, Williams GM, Yang J, Field A, Schultz KA, et al. Nasal chondromesenchymal hamartomas arise secondary to germline and somatic mutations of DICER1 in the pleuropulmonary blastoma tumor predisposition disorder. Hum Genet. 2014;133:1443–50.
Koelsche C, Mynarek M, Schrimpf D, Bertero L, Serrano J, Sahm F, et al. Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathol. 2018;136:327–37.
Schultz KAP, Nelson A, Harris AK, Finch M, Field A, Jarzembowski JA, et al. Pleuropulmonary blastoma-like peritoneal sarcoma: a newly described malignancy associated with biallelic DICER1 pathogenic variation. Mod Pathol. 2020. https://doi.org/10.1038/s41379-020-0558-4.
Nie X, Wang Q, Jiao K. Dicer activity in neural crest cells is essential for craniofacial organogenesis and pharyngeal arch artery morphogenesis. Mech Dev. 2011;128:200–7.
Gradus B, Alon I, Hornstein E. miRNAs control tracheal chondrocyte differentiation. Dev Biol. 2011;360:58–65.
Barter MJ, Tselepi M, Gomez R, Woods S, Hui W, Smith GR, et al. Genome-wide MicroRNA and gene analysis of mesenchymal stem cell chondrogenesis identifies an essential role and multiple targets for miR-140-5p. Stem Cells. 2015;33:3266–80.
Hettmer S, Archer NM, Somers GR, Novokmet A, Wagers AJ, Diller L, et al. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer. 2014;120:1068–75.
Bardeesy N, Falkoff D, Petruzzi MJ, Nowak N, Zabel B, Adam M, et al. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet. 1994;7:91–7.
Frank AJ, Hernan R, Hollander A, Lindsey JC, Lusher ME, Fuller CE, et al. The TP53-ARF tumor suppressor pathway is frequently disrupted in large/cell anaplastic medulloblastoma. Brain Res Mol Brain Res. 2004;121:137–40.
Wegert J, Vokuhl C, Ziegler B, Ernestus K, Leuschner I, Furtwangler R, et al. TP53 alterations in Wilms tumour represent progression events with strong intratumour heterogeneity that are closely linked but not limited to anaplasia. J Pathol Clin Res. 2017;3:234–48.
Kodet R, Newton WA Jr., Hamoudi AB, Asmar L, Jacobs DL, Maurer HM. Childhood rhabdomyosarcoma with anaplastic (pleomorphic) features. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993;17:443–53.
Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013;24:710–24.
Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. 2015;20:227–43.
Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun. 2015;6:7557.
Lewis N, Soslow RA, Delair DF, Park KJ, Murali R, Hollmann TJ, et al. ZC3H7B-BCOR high-grade endometrial stromal sarcomas: a report of 17 cases of a newly defined entity. Mod Pathol. 2018;31:674–84.
Chiang S, Lee CH, Stewart CJR, Oliva E, Hoang LN, Ali RH, et al. BCOR is a robust diagnostic immunohistochemical marker of genetically diverse high-grade endometrial stromal sarcoma, including tumors exhibiting variant morphology. Mod Pathol. 2017;30:1251–61.
Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, et al. Associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res. 2018;24:2251–61.
de Kock L, Terzic T, McCluggage WG, Stewart CJR, Shaw P, Foulkes WD, et al. DICER1 mutations are consistently present in moderately and poorly differentiated Sertoli–Leydig cell tumors. Am J Surg Pathol. 2017;41:1178–87.
Rabban JT, Karnezis AN, Devine WP. Practical roles for molecular diagnostic testing in ovarian adult granulosa cell tumour, Sertoli–Leydig cell tumour, microcystic stromal tumour and their mimics. Histopathology. 2020;76:11–24.
Acknowledgements
The authors would like to thank the University of Chicago Human Tissue Resource Center for slide preparation and the Molecular Diagnostic Laboratories for performing next-generation sequencing. We would also like to thank Anne-Laure Chong at McGill University for assisting with the laboratory work on case 21.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
Concept and design: JAB, ZO, EO. Case contribution: JAB, RHY, AP, KVDV, EB, RS, WGM, EO. Pathology review: JAB, ZO, RHY, EO. Bioinformatics processing: PW. Molecular analysis and interpretation: ZO, LLR, LDK, WDF. Statistical analysis: JAB. Manuscript draft: JAB, EO. Review and editing of manuscript: All authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
This study was approved by the Instutional Review Boards at the individual institutions.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bennett, J.A., Ordulu, Z., Young, R.H. et al. Embryonal rhabdomyosarcoma of the uterine corpus: a clinicopathological and molecular analysis of 21 cases highlighting a frequent association with DICER1 mutations. Mod Pathol 34, 1750–1762 (2021). https://doi.org/10.1038/s41379-021-00821-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00821-x
This article is cited by
-
Mullerian adenosarcoma: clinicopathologic and molecular characterization highlighting recurrent BAP1 loss and distinctive features of high-grade tumors
Modern Pathology (2022)
-
DICER1-sarcoma: an emerging entity
Modern Pathology (2021)
