
Key Points
-
Testicular germ cell tumour (TGCT) has notably high heritability with commensurate high familial relative risks compared with other cancers
-
Genome-wide association studies have identified 25 loci for TGCT, consistent with a highly polygenic architecture underlying the genetic susceptibility
-
Intratubular germ cell neoplasia unclassified, the noninvasive precursor of TGCT, is molecularly similar to primordial germ cells and likely arises during fetal development, lying dormant in the gonads until puberty
-
Testicular tumours are mutationally stable and are characterized by KIT and KRAS mutations at modest frequency. Large-scale chromosomal gains are common in testicular tumours. Nearly all tumours carry isochromosome 12p, which is likely to be a key triggering event for malignant transformation
-
Platinum resistance affects <10% of patients but results in poor outcomes; although extensively investigated through somatic and germline pharmacogenomic studies, the mechanism remains obscure
Abstract
The genomic landscape of testicular germ cell tumour (TGCT) can be summarized using four overarching hypotheses. Firstly, TGCT risk is dominated by inherited genetic factors, which determine nearly half of all disease risk and are highly polygenic in nature. Secondly KIT–KITLG signalling is currently the major pathway that is implicated in TGCT formation, both as a predisposition risk factor and a somatic driver event. Results from genome-wide association studies have also consistently suggested that other closely related pathways involved in male germ cell development and sex determination are associated with TGCT risk. Thirdly, the method of disease formation is unique, with tumours universally stemming from a noninvasive precursor lesion, probably of fetal origin, which lies dormant through childhood into adolescence and then eventually begins malignant growth in early adulthood. Formation of a 12p isochromosome, a hallmark of TGCT observed in nearly all tumours, is likely to be a key triggering event for malignant transformation. Finally, TGCT have been shown to have a distinctive somatic mutational profile, with a low rate of point mutations contrasted with frequent large-scale chromosomal gains. These four hypotheses by no means constitute a complete model that explains TGCT tumorigenesis, but advances in genomic technologies have enabled considerable progress in describing and understanding the disease. Further advancing our understanding of the genomic basis of TGCT offers a clear opportunity for clinical benefit in terms of preventing invasive cancer arising in young men, decreasing the burden of chemotherapy-related survivorship issues and reducing mortality in the minority of patients who have treatment-refractory disease.
Similar content being viewed by others


Main
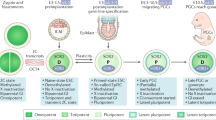
Testicular germ cell tumour (TGCT) is the most common malignancy affecting young men, with a mean age at diagnosis of 36 years1,2. In Western industrialised nations the incidence of TGCT has approximately doubled over the last 4 decades3, providing strong motivation to understand its biological and genetic basis. Cure rates for TGCTs are typically high, owing to the effectiveness of platinum-based chemotherapy, but survivorship comes with a burden of long-term sequelae including increased risk of metabolic syndrome, infertility and secondary cancer4,5,6. Furthermore, limited options exist for patients whose cancer demonstrates platinum resistance, for whom 5-year survival is only 10–15%7. The advent of genome-wide array and next-generation sequencing technologies has heralded rich insights into TGCT oncogenesis, revealing genetic determinants of the strong inherited risk, as well as a distinctive somatic mutational profile. In this Review we discuss these findings and outline the emerging genomic architecture of TGCT using a timeline approach (Fig. 1), as well as highlighting the potential implications for disease treatment and drug resistance.

CNV, copy number variation; FRR, familial relative risk; i(12p), 12p isochromosome; ITGCN, intratubular germ cell neoplasia unclassified; m, mutant; TGCT, testicular germ cell tumour; wt, wild-type.
Before cancer develops
Predisposition to TGCT
Evidence from family, twin and migration studies support a strong inherited genetic basis to TGCT susceptibility, with brothers of patients having an estimated fourfold to tenfold increased risk8,9,10,11. A population-wide analysis of the Swedish family cancer database published in 2015 has estimated the heritability of TGCT to be 48.9%12, suggesting that nearly half of all TGCT risk is determined by inherited genetic factors. These values suggest a substantially higher component of genetic susceptibility than common tumour types such as breast or prostate cancer, for which sibling relative risk is typically approximately twofold increased and heritability in the region of 30%13,14. Consistent with a strong inherited genetic component, disease incidence also varies considerably between ethnic groups, with white American men being at a fivefold higher risk than their African-American counterparts, despite several generations since migration9. Elucidation of these genetic risk factors was first approached through genome-wide linkage analysis, which essentially yielded null results, indicating that a single high-penetrance risk locus is unlikely to exist15,16. Through subsequent candidate association studies a 1.6 Mb deletion on chromosome Y (designated gr/gr) was identified, which confers a twofold elevation in TGCT risk17. However, the frequency of the gr/gr deletion is low (observed in ∼2% of cases), meaning it accounts for only ∼0.5% of the total genetic (excess familial) risk of TGCT development. Furthermore, whole-exome sequencing, which has been undertaken in >900 TGCT patients including >150 multiplex TGCT families has also, thus far, failed to identify high-risk TGCT predisposition genes of significant frequency (C. Turnbull, R. Houlston, R. Giles unpublished data and personal communication). However, coding variants conferring intermediate-to-high risk of TGCT might still exist, but they are likely to each only account for a small proportion of cases of multiple occurrence TGCT families. A number of additional strands of evidence also support an alternative, highly polygenic model of TGCT susceptibility, with disease risk determined by the co-inheritance of multiple risk variants, many of which are common18. Heritability estimates using genomic datasets suggest that over three-quarters of the inherited genetic risk is transmitted through common variation12. Exposition of this common component has come from genome-wide association studies (GWAS), which have so far identified 25 risk loci for TGCT19,20,21,22,23,24,25,26,27,28 (Table 1). Each individual common variant only makes a modest contribution to the genetic risk of TGCT, but notably the TGCT risk single nucleotide polymorphisms (SNPs) identified to date include individual SNPs carrying per-allele odds ratios in excess of 2.6, which is among the highest reported in GWAS of any disease phenotype29.
In addition to high effect sizes, the associations identified by GWAS have provided novel biological insights into the development of TGCT, highlighting in particular genes related to KIT–KITLG signalling, other pathways of male germ cell development, sex determination and genomic integrity (Fig. 2, Table 1). The first risk locus was identified at 12q21, which contains KITLG, and this locus remains the strongest genetic risk factor for TGCT (per-allele OR >2.6). KITLG encodes the ligand for the membrane-bound receptor tyrosine kinase mast/stem cell growth factor receptor Kit (KIT), which regulates the survival, proliferation and migration of germ cells30. Prior to discovery of this locus by GWAS, KIT signalling had already been strongly implicated in TGCT, with mice null for either Kit or Kitlg being shown to be infertile31 and mice with a heterozygous deletion encompassing just the Kitlg region having a twofold elevated risk of developing TGCT32. Functional studies in human cell lines have elucidated that the signal of association at this locus is mediated through an allele-specific p53 binding effect and subsequent upregulation of KITLG expression, which likely increases KIT signalling and proliferation of germ cells33. Analysis of a GWAS identified three other risk loci containing genes related to KIT: 5q31 (SPRY4), 6p21 (BAK1) and 11q14.1 (GAB2). Sprouty homologue 4 (SPRY4) inhibits the mitogen-activated protein kinase (MAPK3–MAPK1) pathway, which is in turn activated by KIT signalling34. The expression of apoptosis-promoting BAK1 is also regulated by KIT35. GAB2, a member of the GRB2-associated binding protein (GAB) family, also associates with KIT and forms a critical part of this KIT–KITLG signalling cascade36. GWAS together with wide-ranging functional evidence have established the KIT pathway as a central tenet in TGCT formation; not only is it activated during overt tumorigenesis but it is also involved in predefining disease risk.

Identified using Search Tool for the Retrieval of Interacting Genes/Proteins database127 with genes implicated in TGCT risk loci and/or development highlighted with a red outline.
Other pathways of male germ cell development have also been repeatedly highlighted in TGCT GWAS, including genes at 3p24.2 (DAZL) and 8q13.3 (PRDM14). DAZL encodes an RNA-binding protein that has been shown to have a central role in the early differentiation of human primordial germ cells (PGCs) in expression studies: Dazl−/− mice are infertile, with differentiation of the germ cells halted at the type A spermatogonia stage37. PRDM14 encodes a transcriptional regulator and modulates PGC specification through control of expression of key pluripotency genes, such as POU5F1 (also known as OCT4), NANOG and SOX2 (Ref. 38). A distinct but closely related pathway also repeatedly highlighted in GWAS is sex determination, in particular at locus 9p24, which contains a single gene, DMRT1. In many species, high expression of DMRT1 is required for differentiation along the male lineage39; furthermore, DMRT1 deficiency is also associated with testicular cancer in mouse models: 90% of Dmrt1−/− 129Sv mice develop teratomas compared with <1% of Dmrt1+/+ mice40. Another gene set repeatedly implicated in TGCT risk is those genes involved in the centrosome cycle and microtubule assembly, with loci containing CENPE, TEX14, PMF1 and MAD1L1 showing association in GWAS studies41. Other biological pathways putatively associated with TGCT by GWAS relate to genomic integrity, such as telomerase function and DNA damage repair. However, although many of the genes and pathways implicated in TGCT through GWAS have extensive pre-existing evidence to support their role in TGCT formation, a GWAS signal points only to a genomic block of linkage disequilibrium. Hence, further functional fine mapping is urgently required to validate and expand this putative attribution of signals to particular genes.
The current known TGCT risk loci collectively explain ∼25% of the excess sibling risk of TGCT and (Fig. 3; Table 1), accordingly, multiple additional TGCT susceptibility loci likely exist and remain to be identified. This notion is supported by a genome-wide complex trait analysis42 conducted for TGCT12, which suggested that at least 50 additional risk SNPs with an odds ratio of ∼1.2 are likely to exist. Or, more plausibly, with a trailing set of effect sizes (OR = 1.01–1.20), the undiscovered set could be considerably larger12. Thus, the prevailing evidence supports a genomic architecture of TGCT predisposition dominated by multiple common risk loci, perturbing a consistent set of biological pathways.

In addition to genetic predisposition, research into environmental risk factors has been of increasing interest, on account of the rapidly rising incidence of TGCT. Many hypotheses have been explored concerning maternal and in utero exposures: to date, no compelling evidence has been reported implicating specific environmental risk factors in the aetiology of TGCT43. The rise in TGCT incidence has been accompanied by a concordant fall in global sperm counts, prompting the hypothesis that TGCT might be aetiologically linked to male reproductive abnormalities as a part of the so-called 'testicular dysgenesis syndrome' (TDS)44. Indeed, genetic studies have found shared risk factors, such as the chromosome Y gr/gr deletion, which predisposes to infertility as well as TGCT17. A large meta-analysis showed that subfertile men have a 1.6-fold increased risk of TGCT and a Danish study reviewing >200 contralateral biopsy specimens in men after a first TGCT showed evidence of testicular dysgenesis in 25% of samples. Other recognized risk factors for TGCT include a history of undescended testis (UDT) and other testicular abnormalities such as hypogonadism and microlithiasis45,46. Elevated TGCT risk is also associated with Down syndrome (trisomy 21)47 and Klinefelter syndrome (47XXY karyotype)48 and, intriguingly, somatic gains of both chromosomes 21 and X are frequently observed in TGCTs49. Furthermore, co-occurrence in families of ovarian germ cell tumour and TGCT has also been recurrently reported, suggesting shared oncogenic pathways50. Finally, epigenetic influences on TGCT predisposition have attracted widespread interest, owing to both the known vulnerability of epigenetic reprogramming to environmental exposures and the established role of DNA methylation in the formation of many cancer types51. Proof of concept for this phenomenon has been demonstrated at locus 5q31 (SPRY4) in analysis of a Scandinavian dataset, with a strong parent-of-origin effect, with TGCT risk associated with maternal (OR = 1.72) but not paternal (OR = 0.99) transmission of the risk allele52. This result is consistent with an imprinting effect, whereby epigenetic mechanisms control gene expression and risk is silenced in paternally inherited alleles.
Origin of the tumour
TGCT is generally accepted to arise from a noninvasive precursor lesion termed intratubular germ cell neoplasia unclassified (ITGCN, formerly known as carcinoma in situ, CIS) that is located in the spermatogonial niche of the seminiferous tubule of the adult human testis53. Molecular observations of ITGCN suggest close phenotypic similarities to PGCs, or gonocytes, with the possibility that normal germ cells transform into ITGCN during fetal development54,55,56. This model is consistent with the clinical finding of ITGCN in a limited number of second trimester fetuses57. The incidence of ITGCN is equivalent to the lifetime risk of developing TGCT, which combined with data from longitudinal studies, has led to wide acceptance that ITGCN progresses to invasive TGCT in most patients.
Expression of pluripotency factors POU5F1 (also known as OCT3/4) and NANOG together with imprinting patterns characterize ITGCN cells as the malignant equivalent of PGCs and gonocytes. The genetic features of ITGCN have not been rigorously profiled; however, targeted sequencing studies have documented examples of KIT mutation, which are well described in TGCT58. Following a primary TGCT, men have a 12-fold elevated risk of disease in the contralateral testicle59. A pertinent question is whether these bilateral tumours originate from a common preinvasive source or whether other predisposing mechanisms cause multiple independent TGCTs. In this context, the identification of concordant KIT mutations in bilateral TGCTs was initially proposed as evidence of a common precursor lesion with mutation occurring during early fetal development58; however, in larger subsequent studies this pattern was not replicated60. The question of mutational concordance in bilateral disease and tumour origin remains a compelling line of inquiry, with the potential to substantially increase our understanding of TGCT aetiology.
Progression to cancer
Tumour subtypes
The model that preinvasive ITGCN cells lie dormant in the gonads during fetal development, infancy and childhood is widely accepted. Progression towards invasive TGCT then occurs after puberty, when ITGCN cells begin to proliferate, likely owing to hormonal influences61,62. The presence of ITGCN can be ascertained via testicular biopsy. Following detection of ITGCN, invasive TGCT is observed in 50% of patients within 5 years, 70% within 7 years and, eventually, in almost all patients63. ITGCN transforms into two main TGCT histological subtypes: seminomas, which resemble undifferentiated PGCs, and nonseminomas, which show differing degrees of differentiation. Seminomas generally arise later in life, with a mean age at diagnosis of 35 years, compared with 25 years for nonseminomas. Transcriptional profiling has identified differing gene signatures for each subtype, with spermatogenesis-associated genes (such as PRAME, MAGEA4 and SPAG1) overexpressed in seminomas and regulatory genes (DNMT3B and SOX2) overexpressed in nonseminoma subtypes64,65. However, despite these morphological differences, several observations suggest commonality of underlying oncogenic pathways and that histological differences are likely to develop late in the tumour formation. For example, 15% of TGCTs have a mixed pathology66 and discordant histologies are frequently reported in both bilateral and familial cases67,68. In addition, GWAS results, when stratified by histology, have been noteworthy for their consistent lack of difference in genetic risk between seminoma and nonseminoma, despite sufficient power for detection19,22,24.
Clinical presentation
Somatic mutational profile of TGCTs. Study of TGCT has revealed that mutational profiles are similar between tumour subtypes, with a markedly low rate of somatic mutation. In fact whole-exome sequencing studies published in 2015 (Refs 49,69) have reported a mean rate of 0.5 somatic mutations per megabase for TGCT, a rate considerably lower than other adult solid tumours such as lung cancer (8.0 mutations/Mb) and melanoma (11.0 mutations/Mb). Overall the TGCT mutation rate is observed to be eightfold lower than the average value observed across multiple cancer types (reported as 4.0/Mb across 27 tumour types70), and only marginally greater than paediatric cancers such as Ewing sarcoma (0.3 mutations/Mb) and rhabdoid tumour (0.15 mutations/Mb). This low mutational frequency is consistent with the hypothesized embryonic origin of TGCT54. The most frequently mutated TGCT driver gene is KIT, in which activating mutations are observed in 25–30% of seminomas71,72. Mutations are found to cluster in the juxtamembrane and kinase-encoding domains of KIT71,72, with by far the most frequent mutations being D816V and D816H. KIT mutation is also associated with other cancer types including myeloid leukaemia73. In addition to KIT, mutations in KRAS have consistently been reported, albeit at a lower frequency of ∼5–10%. Identification of KIT and KRAS mutations was through initial targeted and hypothesis-driven analyses of individual genes and gene sets, which led to subsequent whole-exome profiling studies; however no driver genes with higher mutational frequency have been reported in these subsequent nontargeted studies. Several lower frequency driver-gene candidates have been proposed; however, further large-scale sequencing studies (>300 tumours) will be required to robustly assess these candidates. TGCTs are also noted for their general absence of TP53 mutation, with wild-type TP53 retained in >97% of testicular tumours, compared with only ∼50% of all cancers overall74,75,76. However, TP53 signalling might still be disrupted in TGCTs via indirect mechanisms such as overexpression of MDM2 or the activity of microRNAs (miR) such as miR-372 and miR-373 (Refs 77,78).
In contrast to the low rate of point mutations, TGCTs are highly aneuploid, with large-scale copy number variations (CNV) frequently observed. This aneuploidy is proposed to be derived from an initial tetraploidization event followed by chromosome loss79. Gain of chromosomal material from 12p is observed in almost all tumours, and most commonly manifests as a 12p isochromosome, known as i(12p)80,81,82. i(12p) is widely regarded as the hallmark of TGCT and minimal mapping of the 12p region involved in cases without an i(12p), undertaken to identify a causal driver gene or driver genes, has been described in several studies83,84. Several genes have been proposed as candidates driving the selection for i(12p), including KRAS at 12p11.2-p12.1 and a cluster of stem-cell-associated genes such as NANOG and DPPAS (also known as STELLAR) at 12p13.31, all of which are overexpressed in TGCTs83,84. However, definitive evidence is still lacking as to both the exact driver genes and functional mechanisms underlying i(12p). In addition to minimal mapping, the study of i(12p) in precursor ITGCN cells has been of considerable interest, with the majority of studies demonstrating that premalignant ITGCN (those with no adjacent invasive TGCT) does not contain i(12p), but malignant ITGCN does85,86. Hence, it is hypothesised that i(12p) is not required for ITGCN formation, but is the triggering event that is responsible for driving invasive growth. Overall, presence of i(12p) is widely accepted as critical in TGCT pathogenesis.
Recurring gain of chromosomes 7, 8, 21, 22 and X and loss of chromosome Y are also consistently reported at a frequency of 25–40% TGCTs49,80,81,82,87,88,89,90. In addition to these large-scale CNVs (>1Mb), specific amplification of the KIT gene at 4q12 leads to overexpression and is an alternative mechanism to activating mutations that enhances KIT signalling in seminomas71. Two large independent studies have sought to further profile focal CNVs (<1Mb) in TGCTs49,91 with both studies reporting a recurrent small-scale focal gain at 2q32.1, encompassing gene FSIP2, present in 15–20% of each of the independent TGCT cohorts. FSIP2 encodes a protein that forms part of the fibrous sheath, a cytoskeletal structure located in the principal piece region of the sperm flagellum. FSIP2 expression is testis-specific92 and it is hypothesized to act as a linker protein, binding AKAP4 to the fibrous sheath92. Dysplasia of the fibrous sheath and mutations in AKAP4 have both been linked to male infertility93,94. Aside from 2q32.1, these studies of focal CNVs also identified a number of other candidate regions with recurring focal CNV, which, conversely, failed to be replicated across studies, indicative of imperfect sensitivity and/or specificity of the copy-number calling and/or lack of power of these studies.
Finally, the somatic mutational profile of TGCTs show strong similarities across histological subtypes, with almost identical mutation rates in both seminomas and nonseminomas (0.50 and 0.49 mutations per megabase, respectively) and i(12p) being a typical feature of both49. Two consistent differences exist between the subtypes: firstly, KIT mutations are found to be largely unique to seminomas, and, secondly, CNV profiles show that seminomas are hypertriploid, whereas nonseminomas are hypotriploid. Additional subtle differences between the subtypes have been reported including differential loss of heterozygosity rates, but larger in-depth studies are required for further clarification95. Another clinically associated feature of note is a correlation between the somatic mutational rate and patient age, with the rate increasing with age. This observation suggests that the majority of somatic TGCT mutations are passengers that simply accumulate with patient age, following their early initiation of the disease.
During and after treatment
Response to chemotherapy
Substantial advances in the treatment of TGCT have been made, owing to the exceptional sensitivity of malignant testicular germ cells to platinum-based chemotherapies. Today, a cure is expected in >95% of all patients and in ∼80% of patients presenting with metastatic disease96,97. Despite these high cure rates, a minority of tumours exhibit resistance to platinum therapy: treatment options for these men are limited and long-term survival prospects are poor. Indeed, the average age at death for these men is 32 years; hence, the identification of novel treatment options that are effective in the platinum-refractory setting is an urgent priority. Several hypotheses have been proposed to explain both the exceptional sensitivity of TGCTs to platinum as well as the corresponding mechanisms of resistance. The overall low somatic mutation rate of TGCTs is likely to be a contributing factor to the hypersensitive chemoresponse, with a reduced genetic diversity and decreased probability of pre-existing mutation in a resistance-driving gene. Another model proposes that treatment sensitivity is caused by the inability of TGCTs to repair treatment-induced DNA damage, owing to the low expression of DNA repair genes such as ERCC1 (Ref. 98). Conversely, resistant TGCTs might acquire an enhanced capacity to repair DNA and avoid apoptosis. Notably, sensitivity to poly(ADPribose) polymerase (PARP) inhibition has been observed in platinum-resistant TGCT cell lines, suggesting these cells have deficiency in homologous recombination pathways99. An additional feature, which might partly explain TGCT platinum sensitivity, is that — unlike most other solid tumours — TP53 is rarely mutated in TGCTs. In fact, p53 expression is not only retained at normal levels in TGCTs but is frequently upregulated, which is considered to be a major factor in explaining platinum hypersensitivity100. A variety of other models have also been proposed, including a hypersensitive apoptotic response to platinum or, alternatively, epigenetic mechanisms whereby sensitivity is related to the hypomethylation status of some TGCT genomes101.
Clinical studies of platinum resistance have also been performed, comparing treatment-sensitive with treatment-resistant tumours. Initial research highlighted mutation of BRAF, with the V600E variant present in 26% of resistant tumours versus 1% of sensitive tumours102; however, this finding was not replicated in larger subsequent studies103. Hotspot mutational analysis identified PIK3CA and/or AKT1 variants in 7% of resistant tumours (n = 46), versus 0% in samples from sensitive tumours (n = 24), as well as an excess of RAS, and FGFR3 mutations in resistant tumours103. Whole-exome sequencing of two treatment-resistant tumours identified a missense mutation in DNA repair gene XRCC2 (Ref. 49), suggesting activation of DNA repair pathways in response to treatment-induced damage. Interestingly, mutation of the related DNA repair gene ERCC2 has been found to correlate with platinum response in a exome-wide study of 50 urothelial carcinomas104. Aside from XRCC2 mutation, the exome profile of resistant TGCT seems to be comparable with sensitive tumours, with a consistent mutational load of 0.5 mutations per megabase, albeit based on only two samples. The role of KIT has also been studied in treatment response; however, a clinical trial of imatinib (which inhibits activated KIT) showed no antitumour activity against KIT mutation-positive platinum-refractory TGCTs105. CCND1 overexpression has also been associated with cisplatin resistance in TGCTs as well as other tumour types106,107. Platinum resistance has also been explored in several germline pharmacogenomics studies, with polymorphism of the gene PAI-1 (also known as SERPINE1) being shown to act as a predictor of negative platinum response, with homozygote risk-allele carriers having a hazard ratio of 2.69 for TGCT-related death108. However, owing to the difficulty in obtaining large numbers of samples for which both DNA and clinical data on treatment response are available, the majority of germline biomarkers have not yet been validated in large cohorts. Thus, the mechanisms driving platinum resistance remain unclear and are likely to involve a sequence of genetic and/or epigenetic changes, delineation of which will be critical to identifying improved treatment options and prognostic biomarkers.
After cancer
Long-term survivorship issues
The success of treating TGCT has been accompanied by an increase in survivorship issues, caused by the long-term consequences associated with platinum-based chemotherapy. A number of associated morbidities have been identified, including neurotoxicity, hypogonadism, infertility and increased risks of both cardiovascular disease and secondary cancer4,5,6,109. For example neuropathic symptoms are reported in 20% of TGCT survivors at 2 years after combination chemotherapy, and cisplatin treatment is associated with ototoxicity (high-frequency hearing loss and tinnitus). Genetic studies have reported polymorphisms associated with predisposition to these adverse effects, such as variations in GSTP1, which are associated with an increased risk of peripheral neuropathy and ototoxicity110,111. In terms of hypogonadism, a proportion of patients will already have low testosterone levels before TGCT (through TDS), whereas chemotherapy and radiotherapy have been shown to increase hypogonadism rates from 11% to 38%112. In addition, the deleterious effects of chemotherapy on spermatogenesis have been well documented5, with 20–30% of men being left infertile after TGCT treatment106. Moreover, even in men whose sperm counts do recover, sperm DNA damage after chemotherapy can persist 2 years after treatment, compromising gamete quality and causing potential risks for offspring113. Finally, both cardiovascular and secondary cancer risks are elevated following chemotherapy, with an increased incidence of metabolic syndrome (central obesity, dyslipidaemia and hypertension) and secondary leukaemia in particular. A large-scale study of >40,000 TGCT survivors demonstrated a lifetime risk of secondary cancer of 31% for patients with seminoma and 36% for men with nonseminoma, respectively, compared with 23% for the general population114.
Other clinical applications of genetics
Currently, no clinical application of genetics exists for predicting disease or stratifying clinical care, but a number of clinically orientated studies are underway to explore its potential utility. Disease prevention has been a central focus, with the large effect sizes of TGCT GWAS SNPs suggesting genetic screening could be clinically useful29. Preliminary assessment of genetic profiling has been approached by two groups20,115, who used polygenic-risk-scoring (PRS) models to consider the combined effect of all known SNPs on TGCT risk. The latest such model demonstrates that the top 1% of men with the highest genetic risk of TGCT carry a 10.4-fold elevated disease risk compared with the population average. In comparison, equivalent PRS models for ovarian, breast and prostate cancer, show the top 1% of highest-risk genotypes have only a twofold, threefold and fivefold increased risk, respectively116, indicating that the TGCT SNPs have useful power in terms of risk discrimination. However, the case for screening is weaker than that for breast or prostate cancer, as TGCT has much lower population frequency than these other cancers and has an excellent prognosis. The rare nature of TGCT (lifetime male Caucasian absolute risk of 0.5%) means high relative risks translate into only modest absolute risk: the 1% of men with the highest risk of TGCT have an notable relative risk of 10.4, but still only have a ∼5% lifetime risk of TGCT117. It might be possible to combine genetic and nongenetic risk factors to improve the discriminatory performance of risk profiling. For example, one model testing this approach has predicted that men in the top 1% of genetic risk, and with a history of UDT, would have a relative risk of 50 (Ref. 115), equating to a lifetime TGCT risk of ∼25%. However, this model assumes full independence of effects between genetic and nongenetic factors, an assumption not yet validated through the relevant modelling of large datasets combining clinical and genetic data. Currently, men at high risk of TGCT would proceed to bilateral testicular biopsy to assess for the presence of the premalignant ITGCN lesion, proceeding to orchidectomy if detected. The invasive nature of testicular biopsy currently restricts development of TGCT screening programmes. Improvements in sensitivity and specificity of semen assay, an emerging noninvasive diagnostic tool by which presence or absence of ITGCN is detected via staining for fetal germ cell markers, could influence the feasibility of TGCT screening in the future118,119. Such an approach would be taking advantage of the unique characteristic of ITGCN as a faithful precursor lesion of TGCT, clinically detectable from adolescence.
Future directions
Large-scale GWAS are expected to identify multiple additional TGCT risk variants as sample sizes increase. For example, the largest TGCT meta-GWAS to date contains 3,556 patient samples and 13,969 control samples, with a 7.2% power to detect common variants120 (defined as OR >1.25 and minor allele frequency (MAF) >5%). Sample numbers are expected to soon reach 10,000, through large collaborative efforts such as the Testicular Cancer Association Consortium (TECAC). At the level of 10,000 cases, the power to detect common variants will increase to 88% (based again on OR>1.25, MAF>5% and 50,000 control samples). Following these studies, considerably more heritable risk factors for TGCT are likely to be identified, offering a greater potential for personalized risk prediction. In addition, the functional fine mapping of TGCT risk loci will become a major focus, as functional genomic evidence is becoming increasingly important for drug target identification and validation. Indeed, evidence shows that drugs developed for genetically validated targets identified in data sources such as GWASdb and Online Mendelian Inheritance in Man are twice as likely to be successful in clinical development than those not validated121. A wide range of functional mapping techniques are likely to be informative, including targeted resequencing, expression quantitative trait loci analysis, transcription factor binding analysis, chromosome-conformation-capture techniques, methylation profiling and direct genotype manipulation in cell-line models. The utility of these techniques to build an integrated functional view of oncogenic pathways will be critical, and likely to be particularly informative for TGCT, given the strong commonality between historic biological evidence, genetic findings and overlapping conditions such as TDS.
Aside from common SNPs, the influence of rare predisposition variants also remains to be revealed. A single high penetrance “TGCT gene” is unlikely to exist, but rare higher-risk mutations could yet be identified through large-scale next-generation sequencing analyses of families with an increased risk of TGCT. In terms of additional determination of somatic variation in TGCT, comprehensive profiling of 150 TGCTs has been undertaken by The Cancer Genome Atlas (TCGA) project. Exome sequencing data in TCGA project, when meta-analysed together with previous data49, confirmed the low mutation rate and absence of other consistent driver genes except KIT and KRAS122 in this series. Additional studies are underway to comprehensively study the genomics of platinum resistance.
Two main animal models predisposed to developing TGCT have been reported: a zebrafish model carrying bmpr1b mutations that disrupt BMP signalling, and 129Sv-strain mice, with complete loss of Dmrt1, which display high incidence of teratoma40. Both of these models provide relevant biological insight into TGCT development, but their relevance to human tumorigenesis is limited, as genes such as BMPR1B have not been shown to be mutated in human TGCT. In 2013, a new zebrafish model with inactivating mutations in irrc50 (also known as dnaaf1) that codes for a ciliary protein was reported, with >90% penetrance of seminoma, suggesting an intriguing functional link between TGCT and cilia function123. Despite the utility of these animal models and others, no model currently exists to reflect the biology of human nonseminomas, such as embryonal carcinoma or yolk sac tumour. Given that nonseminomas are generally more aggressive, with increased rates of platinum resistance, an urgent need exists for new models representing these subtypes. Future development and use of these animal models will provide novel insights into the underlying molecular mechanisms of TGCT, as well as useful tools to test therapeutic strategies.
Given the exceptional sensitivity of TGCTs to platinum, future therapeutic advances are likely to focus on chemotherapy in combination with other synergistic compounds. For example, a trial has been conducted assessing bevacizumab treatment together with high-dose chemotherapy, exploiting the overexpression of vascular endothelial growth factor (VEGF) in metastatic TGCT124,125. This study demonstrated promising results in otherwise refractory cases despite its small size (n = 37), with an overall survival rate of 58% at 46 months, albeit with 11% of patients dying from treatment-related toxicities125. Further integrated genomic and/or transcriptomic profiling of refractory TGCTs might suggest additional rational treatment combinations of platinum together with other targeted molecules. Immunotherapeutic strategies have been postulated as a long-term strategy, with the additional benefit of reduced toxic effects; a 2015 study showed frequent expression of the programmed death ligand 1 (PD-L1) in TGCTs126. This finding is somewhat counterintuitive, given that a high mutational load is hypothesized to be a prerequisite for an immune-mediated response, as evidenced by the effectiveness of PD-L1 inhibition in highly mutated tumours such as melanoma and lung cancer125. Clearly TGCT, with its low mutation rate, does not fit this same profile; however, increasing evidence now suggests that tumour immune response is mediated by a more complex set of factors125.
Attention will continue to focus on longstanding unresolved questions of TGCT oncogenesis, such as tumour origin and the functional mechanisms of i(12p). With regard to tumour origin, detailed profiling of bilateral tumours from the same patient, together with the ITGCN precursor cells, would comprehensively address the question of common ancestry, although a more immediately tractable approach might come from use of model systems in the first instance. Lastly, insights into the functional significance of i(12p) may be forthcoming through a number of approaches, such as integrated functional analysis of TCGA data, or long-read or whole-genome sequencing to finely map the structure of i(12p), detecting any complex rearrangements or gene fusion events.
Conclusions
In summary, we have described the genomic landscape of TGCT from susceptibility through oncogenesis to therapeutic response and sequelae thereof. TGCT is characterised by its strong and highly polygenic genetic susceptibility. GWAS have identified 25 loci for TGCT, which together explain >25% of its genetic determinants and have implicated gene sets relating to specific pathways such as KIT–KITLG signalling, transcriptional regulation of primordial male germ cell development, sex determination and centrosome cycle-microtubule assembly. Large-scale whole-exome sequencing has confirmed that TGCTs are quiet with regard to point mutations, characterized only by recurrent mutation at modest frequency of KIT and KRAS, but that large-scale chromosomal gains are frequent, with i(12p), which is observed in nearly all tumours, being the likely driver event of malignant transformation. Platinum resistance, affecting <10% of patients, is the subject of multiple somatic and germline pharmacogenomic studies, but, to date, remains poorly inderstood. Further study of genetic susceptibility will likely comprise ever larger GWAS studies, advances in population risk stratification, functional genomic exposition of GWAS loci and study of rare variants through whole-exome and whole-genome sequencing. Further study of the tumours will probably explore the biology of tumour development including the architecture of the i(12p). TGCT is a cancer of comparatively low frequency that is typically platinum sensitive, but additional exploration of the genomic basis of TGCT remains a research priority offering clinical benefit, with opportunity for cancer prevention, reduction in chemotherapy-related survivorship issues in young men and reducing mortality in those men with treatment-refractory disease.
References
Bray, F., Ferlay, J., Devesa, S. S., McGlynn, K. A. & Moller, H. Interpreting the international trends in testicular seminoma and nonseminoma incidence. Nat. Clin. Pract. Urol. 3, 532–543 (2006).
Ruf, C. G. et al. Changes in epidemiologic features of testicular germ cell cancer: age at diagnosis and relative frequency of seminoma are constantly and significantly increasing. Urol. Oncol. 32, 33.e1–33.e6 (2014).
Le Cornet, C. et al. Testicular cancer incidence to rise by 25% by 2025 in Europe? Model-based predictions in 40 countries using population-based registry data. Eur. J. Cancer 50, 831–839 (2014).
de Haas, E. C. et al. Early development of the metabolic syndrome after chemotherapy for testicular cancer. Ann. Oncol. 24, 749–755 (2013).
Bujan, L. et al. Impact of chemotherapy and radiotherapy for testicular germ cell tumors on spermatogenesis and sperm DNA: a multicenter prospective study from the CECOS network. Fertil. Steril. 100, 673–680 (2013).
Rusner, C. et al. Risk of second primary cancers after testicular cancer in East and West Germany: a focus on contralateral testicular cancers. Asian J. Androl. 16, 285–289 (2014).
Nitzsche, B. et al. Anti-tumour activity of two novel compounds in cisplatin-resistant testicular germ cell cancer. Br. J. Cancer 107, 1853–1863 (2012).
Swerdlow, A. J., De Stavola, B. L., Swanwick, M. A. & Maconochie, N. E. Risks of breast and testicular cancers in young adult twins in England and Wales: evidence on prenatal and genetic aetiology. Lancet 350, 1723–1728 (1997).
McGlynn, K. A., Devesa, S. S., Graubard, B. I. & Castle, P. E. Increasing incidence of testicular germ cell tumors among black men in the United States. J. Clin. Oncol. 23, 5757–5761 (2005).
Hemminki, K. & Li, X. Familial risk in testicular cancer as a clue to a heritable and environmental aetiology. Br. J. Cancer 90, 1765–1770 (2004).
Kharazmi, E. et al. Cancer risk in relatives of testicular cancer patients by histology type and age at diagnosis: a joint study from five Nordic countries. Eur. Urol. 68, 283–289 (2015).
Litchfield, K. et al. Quantifying the heritability of testicular germ cell tumour using both population-based and genomic approaches. Sci. Rep. 5, 13889 (2015).
Locatelli, I., Lichtenstein, P. & Yashin, A. I. The heritability of breast cancer: a Bayesian correlated frailty model applied to Swedish twins data. Twin Res. 7, 182–191 (2004).
Kampman, E. A first-degree relative with colorectal cancer: what are we missing? Cancer Epidemiol. Biomarkers Prev. 16, 1–3 (2007).
Crockford, G. P. et al. Genome-wide linkage screen for testicular germ cell tumour susceptibility loci. Hum. Mol. Genet. 15, 443–451 (2006).
Rapley, E. A. et al. Localization to Xq27 of a susceptibility gene for testicular germ-cell tumours. Nat. Genet. 24, 197–200 (2000).
Nathanson, K. L. et al. The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am. J. Hum. Genet. 77, 1034–1043 (2005).
Pathak, A. et al. Prospectively identified incident testicular cancer risk in a familial testicular cancer cohort. Cancer Epidemiol. Biomarkers Prev. 24, 1614–1621 (2015).
Rapley, E. A. et al. A genome-wide association study of testicular germ cell tumor. Nat. Genet. 41, 807–810 (2009).
Turnbull, C. & Rahman, N. Genome-wide association studies provide new insights into the genetic basis of testicular germ-cell tumour. Int. J. Androl. 34, e86–e96; discussion e96–e97 (2011).
Kanetsky, P. A. et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat. Genet. 41, 811–815 (2009).
Turnbull, C. et al. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat. Genet. 42, 604–607 (2010).
Kanetsky, P. A. et al. A second independent locus within DMRT1 is associated with testicular germ cell tumor susceptibility. Hum. Mol. Genet. 20, 3109–3117 (2011).
Ruark, E. et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat. Genet. 45, 686–689 (2013).
Bojesen, S. E. et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 45, 371–384 (2013).
Chung, C. C. et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet. 45, 680–685 (2013).
Litchfield, K. et al. Multi-stage genome wide association study identifies new susceptibility locus for testicular germ cell tumour on chromosome 3q25. Hum. Mol. Genet. 24, 1169–1176 (2015).
Kristiansen, W. et al. Two new loci and gene sets related to sex determination and cancer progression are associated with susceptibility to testicular germ cell tumor. Hum. Mol. Genet. 24, 4138–4146 (2015).
Chanock, S. High marks for GWAS. Nat. Genet. 41, 765–766 (2009).
Boldajipour, B. & Raz, E. What is left behind — quality control in germ cell migration. Sci. STKE 2007, e16 (2007).
Roskoski, R. Jr. Signaling by Kit protein-tyrosine kinase — the stem cell factor receptor. Biochem. Biophys. Res. Commun. 337, 1–13 (2005).
Heaney, J. D., Lam, M. Y., Michelson, M. V. & Nadeau, J. H. Loss of the transmembrane but not the soluble kit ligand isoform increases testicular germ cell tumor susceptibility in mice. Cancer Res. 68, 5193–5197 (2008).
Zeron-Medina, J. et al. A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell 155, 410–422 (2013).
Sasaki, A. et al. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Cell Cycle 2, 281–282 (2003).
Yan, W., Samson, M., Jegou, B. & Toppari, J. Bcl-w forms complexes with Bax and Bak, and elevated ratios of Bax/Bcl-w and Bak/Bcl-w correspond to spermatogonial and spermatocyte apoptosis in the testis. Mol. Endocrinol. 14, 682–699 (2000).
Yu, M. et al. The scaffolding adapter Gab2, via Shp-2, regulates kit-evoked mast cell proliferation by activating the Rac/JNK pathway. J. Biol. Chem. 281, 28615–28626 (2006).
Schrans-Stassen, B. H., Saunders, P. T., Cooke, H. J. & de Rooij, D. G. Nature of the spermatogenic arrest in Dazl-/- mice. Biol. Reprod. 65, 771–776 (2001).
Tsuneyoshi, N. et al. PRDM14 suppresses expression of differentiation marker genes in human embryonic stem cells. Biochem. Biophys. Res. Commun. 367, 899–905 (2008).
Smith, C. A., McClive, P. J., Western, P. S., Reed, K. J. & Sinclair, A. H. Conservation of a sex-determining gene. Nature 402, 601–602 (1999).
Krentz, A. D. et al. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc. Natl Acad. Sci. USA 106, 22323–22328 (2009).
Litchfield, K., Shipley, J. & Turnbull, C. Common variants identified in genome-wide association studies of testicular germ cell tumour: an update, biological insights and clinical application. Andrology 3, 34–46 (2015).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
McGlynn, K. A. & Cook, M. B. Etiologic factors in testicular germ-cell tumors. Future Oncol. 5, 1389–1402 (2009).
Skakkebaek, N. E., Rajpert-De Meyts, E. & Main, K. M. Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects. Hum. Reprod. 16, 972–978 (2001).
Trabert, B., Zugna, D., Richiardi, L., McGlynn, K. A. & Akre, O. Congenital malformations and testicular germ cell tumors. Int. J. Cancer 133, 1900–1904 (2013).
Coffey, J. et al. Testicular microlithiasis as a familial risk factor for testicular germ cell tumour. Br. J. Cancer 97, 1701–1706 (2007).
Nizetic, D. & Groet, J. Tumorigenesis in Down's syndrome: big lessons from a small chromosome. Nat. Rev. Cancer 12, 721–732 (2012).
Hasle, H., Mellemgaard, A., Nielsen, J. & Hansen, J. Cancer incidence in men with Klinefelter syndrome. Br. J. Cancer 71, 416–420 (1995).
Litchfield, K. et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat. Commun. 6, 5973 (2015).
Stettner, A. R. et al. Familial ovarian germ cell cancer: report and review. Am. J. Med. Genet. 84, 43–46 (1999).
Kristensen, D. G., Skakkebaek, N. E., Rajpert-De Meyts, E. & Almstrup, K. Epigenetic features of testicular germ cell tumours in relation to epigenetic characteristics of foetal germ cells. Int. J. Dev. Biol. 57, 309–317 (2013).
Karlsson, R. et al. Investigation of six testicular germ cell tumor susceptibility genes suggests a parent-of-origin effect in SPRY4. Hum. Mol. Genet. 22, 3373–3380 (2013).
Oosterhuis, J. W. & Looijenga, L. H. Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 5, 210–222 (2005).
Kristensen, D. M. et al. Origin of pluripotent germ cell tumours: the role of microenvironment during embryonic development. Mol. Cell. Endocrinol. 288, 111–118 (2008).
Skakkebaek, N. E., Berthelsen, J. G., Giwercman, A. & Muller, J. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int. J. Androl. 10, 19–28 (1987).
Rajpert-De Meyts, E. Developmental model for the pathogenesis of testicular carcinoma in situ: genetic and environmental aspects. Hum. Reprod. Update 12, 303–323 (2006).
Jacobsen, G. K. & Henriques, U. V. A fetal testis with intratubular germ cell neoplasia (ITGCN). Mod. Pathol. 5, 547–549 (1992).
Biermann, K. et al. c-KIT is frequently mutated in bilateral germ cell tumours and down-regulated during progression from intratubular germ cell neoplasia to seminoma. J. Pathol. 213, 311–318 (2007).
Fossa, S. D. et al. Risk of contralateral testicular cancer: a population-based study of 29,515 U.S. men. J. Natl Cancer Inst. 97, 1056–1066 (2005).
Coffey, J. et al. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: report of 220 tumors and review of literature. Genes Chromosomes Cancer 47, 34–42 (2008).
Horwich, A., Shipley, J. & Huddart, R. Testicular germ-cell cancer. Lancet 367, 754–765 (2006).
Rajpert-De Meyts, E. et al. The emerging phenotype of the testicular carcinoma in situ germ cell. APMIS 111, 267–278; discussion 278–279 (2003).
Dieckmann, K. P., Kulejewski, M., Heinemann, V. & Loy, V. Testicular biopsy for early cancer detection — objectives, technique and controversies. Int. J. Androl. 34, e7–e13 (2011).
Almstrup, K. et al. Genome-wide gene expression profiling of testicular carcinoma in situ progression into overt tumours. Br. J. Cancer 92, 1934–1941 (2005).
Biermann, K. et al. Genome-wide expression profiling reveals new insights into pathogenesis and progression of testicular germ cell tumors. Cancer Genomics Proteomics 4, 359–367 (2007).
Gori, S. et al. Germ cell tumours of the testis. Crit. Rev. Oncol. Hematol. 53, 141–164 (2005).
Mai, P. L. et al. The International Testicular Cancer Linkage Consortium: a clinicopathologic descriptive analysis of 461 familial malignant testicular germ cell tumor kindred. Urol. Oncol. 28, 492–499 (2010).
Forman, D. et al. Familial testicular cancer: a report of the UK family register, estimation of risk and an HLA class 1 sib-pair analysis. Br. J. Cancer 65, 255–262 (1992).
Cutcutache, I. et al. Exome-wide sequencing shows low mutation rates and identifies novel mutated genes in seminomas. Eur. Urol. 68, 77–83 (2015).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
McIntyre, A. et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res. 65, 8085–8089 (2005).
Kemmer, K. et al. KIT mutations are common in testicular seminomas. Am. J. Pathol. 164, 305–313 (2004).
Kim, H. J. et al. KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia, especially in the subgroup with RUNX1/RUNX1T1 rearrangement. Ann. Hematol. 92, 163–171 (2013).
Bignell, G. et al. Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes Chromosomes Cancer 45, 42–46 (2006).
Heidenreich, A. et al. Immunohistochemical and mutational analysis of the p53 tumour suppressor gene aml the bcl-2 oncogene in primary testicular germ cell, tumours. APMIS 106, 90–99 (1998).
Laumann, R., Jucker, M. & Tesch, H. Point mutations in the conserved regions of the p53 tumor suppressor gene do not account for the transforming process in the Jurkat acute lymphoblastic leukemia T-cells. Leukemia 6, 227–228 (1992).
Voorhoeve, P. M. et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 124, 1169–1181 (2006).
Li, B., Cheng, Q., Li, Z. & Chen, J. p53 inactivation by MDM2 and MDMX negative feedback loops in testicular germ cell tumors. Cell Cycle 9, 1411–1420 (2010).
Suijkerbuijk, R. F. et al. Overrepresentation of chromosome 12p sequences and karyotypic evolution in i(12p)-negative testicular germ-cell tumors revealed by fluorescence in situ hybridization. Cancer Genet. Cytogenet. 70, 85–93 (1993).
Atkin, N. B. & Baker, M. C. Specific chromosome change, i(12p), in testicular-tumors. Lancet 2, 1349–1349 (1982).
Atkin, N. B. & Baker, M. C. i(12p): specific chromosomal marker in seminoma and malignant teratoma of the testis. Cancer Genet. Cytogenet. 10, 199–204 (1983).
Sandberg, A. A., Meloni, A. M. & Suijkerbuijk, R. F. Reviews of chromosome studies in urological tumors.3. Cytogenetics and genes in testicular tumors. J. Urol. 155, 1531–1556 (1996).
Rodriguez, S. et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2–p12.1. Oncogene 22, 1880–1891 (2003).
Korkola, J. E. et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 66, 820–827 (2006).
Ottesen, A. M. et al. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosomes Cancer 38, 117–125 (2003).
Summersgill, B., Osin, P. S., Lu, Y. J., Huddart, R. & Shipley, J. Chromosomal imbalances associated with carcinoma in situ and associated testicular germ cell tumours of adolescents and adults. Br. J. Cancer 85, 213–219 (2001).
Henegariu, O., Vance, G. H., Heiber, D., Pera, M. & Heerema, N. A. Triple-color FISH analysis of 12p amplification in testicular germ-cell tumors using 12p band-specific painting probes. J. Mol. Med. (Berl.) 76, 648–655 (1998).
Roelofs, H. et al. Restricted 12p amplification and RAS mutation in human germ cell tumors of the adult testis. Am. J. Pathol. 157, 1155–1166 (2000).
Summersgill, B. et al. Molecular cytogenetic analysis of adult testicular germ cell tumours and identification of regions of consensus copy number change. Br. J. Cancer 77, 305–313 (1998).
Zafarana, G. et al. 12p-amplicon structure analysis in testicular germ cell tumors of adolescents and adults by array CGH. Oncogene 22, 7695–7701 (2003).
LeBron, C. et al. Genome-wide analysis of genetic alterations in testicular primary seminoma using high resolution single nucleotide polymorphism arrays. Genomics 97, 341–349 (2011).
Brown, P. R., Miki, K., Harper, D. B. & Eddy, E. M. A-kinase anchoring protein 4 binding proteins in the fibrous sheath of the sperm flagellum. Biol. Reprod. 68, 2241–2248 (2003).
Chemes, H. E., Brugo, S., Zanchetti, F., Carrere, C. & Lavieri, J. C. Dysplasia of the fibrous sheath: an ultrastructural defect of human spermatozoa associated with sperm immotility and primary sterility. Fertil. Steril. 48, 664–669 (1987).
Miki, K. et al. Targeted disruption of the Akap4 gene causes defects in sperm flagellum and motility. Dev. Biol. 248, 331–342 (2002).
Vladusic, T. et al. Histological groups of human postpubertal testicular germ cell tumours harbour different genetic alterations. Anticancer Res. 34, 4005–4012 (2014).
Oldenburg, J. et al. Testicular seminoma and non-seminoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 24, 125–132 (2013).
Siegel, R. et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J. Clinicians 62, 220–241 (2012).
Usanova, S. et al. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol.Cancer 9, 248 (2010).
Cavallo, F. et al. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to cisplatin and poly (ADP-ribose) polymerase inhibition. PLoS ONE 7, e51563 (2012).
Gutekunst, M. et al. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS ONE 6, e19198 (2011).
Sheikine, Y. et al. Molecular genetics of testicular germ cell tumors. Am. J. Cancer Res. 2, 153–167 (2012).
Honecker, F. et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 27, 2129–2136 (2009).
Feldman, D. R. et al. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin. Cancer Res. 20, 3712–3720 (2014).
Van Allen, E. M. et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 4, 1140–1153 (2014).
Einhorn, L. H., Brames, M. J., Heinrich, M. C., Corless, C. L. & Madani, A. Phase II study of imatinib mesylate in chemotherapy refractory germ cell tumors expressing KIT. Am. J. Clin. Oncol. 29, 12–13 (2006).
Noel, E. E. et al. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am. J. Pathol. 176, 2607–2615 (2010).
Garcia-Velasco, A. et al. Biological markers of cisplatin resistance in advanced testicular germ cell tumours. Clin. Transl. Oncol. 14, 452–457 (2012).
de Haas, E. C. et al. Association of PAI-1 gene polymorphism with survival and chemotherapy-related vascular toxicity in testicular cancer. Cancer 116, 5628–5636 (2010).
Singhera, M., Lees, K., Huddart, R. & Horwich, A. Minimizing toxicity in early-stage testicular cancer treatment. Expert Rev. Anticancer Ther. 12, 185–193 (2012).
Oldenburg, J. et al. Association between long-term neuro-toxicities in testicular cancer survivors and polymorphisms in glutathione-s-transferase-P1 and-M1, a retrospective cross sectional study. J. Transl. Med. 5, 70 (2007).
Peters, U. et al. Glutathione S-transferase genetic polymorphisms and individual sensitivity to the ototoxic effect of cisplatin. Anticancer Drugs 11, 639–643 (2000).
Huddart, R. A. et al. Fertility, gonadal and sexual function in survivors of testicular cancer. Br. J. Cancer 93, 200–207 (2005).
O'Flaherty, C., Hales, B. F., Chan, P. & Robaire, B. Impact of chemotherapeutics and advanced testicular cancer or Hodgkin lymphoma on sperm deoxyribonucleic acid integrity. Fertil. Steril. 94, 1374–1379 (2010).
Travis, L. B. et al. Second cancers among 40,576 testicular cancer patients: focus on long-term survivors. J. Natl Cancer Inst. 97, 1354–1365 (2005).
Greene, M. H. et al. Familial testicular germ cell tumors (FTGCT) — overview of a multidisciplinary etiologic study. Andrology 3, 47–58 (2015).
Bahcall, O. Risk prediction and population screening for breast, ovarian and prostate cancers. Nat. Genet. http://dx.doi.org/10.1038/ngicogs.5 (2012).
Litchfield, K. et al. Polygenic susceptibility to testicular cancer: implications for personalised health care. Br. J. Cancer 113, 1512–1518 (2015).
Almstrup, K. et al. Screening of subfertile men for testicular carcinoma in situ by an automated image analysis-based cytological test of the ejaculate. Int. J. Androl 34, e21–e30; discussion e30–e30 (2011).
Hoei-Hansen, C. E. et al. Towards a non-invasive method for early detection of testicular neoplasia in semen samples by identification of fetal germ cell-specific markers. Hum. Reprod. 22, 167–173 (2007).
Skol, A. D., Scott, L. J., Abecasis, G. R. & Boehnke, M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 38, 390–390 (2006).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
Litchfield, K. Abstract 2986: meta-analysis of whole exome sequencing data reveals the mutational spectrum of testicular germ cell tumors. Cancer Res. 75, 2986 (2015).
Basten, S. G. et al. Mutations in LRRC50 predispose zebrafish and humans to seminomas. PLoS Genet. 9, e1003384 (2013).
Fukuda, S. et al. Expression of vascular endothelial growth factor in patients with testicular germ cell tumors as an indicator of metastatic disease. Cancer 85, 1323–1330 (1999).
Nieto, Y. et al. Bevacizumab/high-dose chemotherapy with autologous stem-cell transplant for poor-risk relapsed or refractory germ-cell tumors. Ann. Oncol. http://dx.doi.org/10.1093/annonc/mdv310 (2015).
Fankhauser, C. D. et al. Frequent PD-L1 expression in testicular germ cell tumors. Br. J. Cancer 113, 411–413 (2015).
Szklarczyk, D. et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Litchfield, K. et al. Identification of four new susceptibility loci for testicular germ cell tumour. Nat. Commun. 6, 8690 (2015).
Author information
Authors and Affiliations
Contributions
K.L. and M.L. researched data for the article, R.A.H., J.S. and C.T. contributed to discussion of its contents. K.L. wrote the manuscript and K.L., M.L. and C.T. reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Rights and permissions
About this article

Cite this article
Litchfield, K., Levy, M., Huddart, R. et al. The genomic landscape of testicular germ cell tumours: from susceptibility to treatment. Nat Rev Urol 13, 409–419 (2016). https://doi.org/10.1038/nrurol.2016.107
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrurol.2016.107
This article is cited by
-
Germline stem cells in human
Signal Transduction and Targeted Therapy (2022)
-
DNA replication machinery prevents Rad52-dependent single-strand annealing that leads to gross chromosomal rearrangements at centromeres
Communications Biology (2020)
-
MicroRNA and transcription factor co-regulatory networks and subtype classification of seminoma and non-seminoma in testicular germ cell tumors
Scientific Reports (2020)
-
gr/gr deletion predisposes to testicular germ cell tumour independently from altered spermatogenesis: results from the largest European study
European Journal of Human Genetics (2019)
