
Abstract
The purpose of the study is to explore the causative role of TUBB2B gene mutations in patients with different malformations of cortical development. We collected and evaluated clinical and MRI data of a cohort of 128 consecutive patients (61 females and 67 males) in whom brain MRI had detected a spectrum of malformations of cortical development including polymicrogyria or pachygyria, who were mutation-negative to other possible causative genes. Mutation analysis of the TUBB2B gene was performed. We identified three new TUBB2B mutations in three unrelated patients (3 out of 128; 2.3%) with a diffuse and rather symmetrical cortical abnormality, including diffuse polymicrogyria in two and bilateral regional pachygyria in one. One patient harbored a p.Asp417Asn amino-acid substitution in the C-terminal domain of the protein; one patient a p.Asn256Ser amino-acid substitution in the intermediate domain and one patient a p.Leu117Pro amino-acid substitution in the N-terminal domain. The localization of each mutation within the secondary structure of the β2-tubulin polypeptide suggests that these mutations might alter the proper functions of microtubules. The phenotypic spectrum associated with TUBB2B mutations is wider than previously reported and includes diffuse, symmetric malformations of cortical development.
Similar content being viewed by others
Introduction
Polymicrogyria is a common cortical malformation characterized by an excessive number of abnormally small gyri that result in an irregular cortical surface with lumpy aspect. Brain pathology demonstrates abnormal development or loss of neurons in middle and deep cortical layers, variably associated with an unlayered cortical structure.1, 2 Several genes have been associated with polymicrogyria, including GPR56, SRPX2, TUBB2B, TUBB3, PAX6, TBR2, KIAA1279, NHEJ1, RAB3GAP1 and TUBA8 2, 3, 4 with all but GPR56, TUBB3 and TUBB2B found in rare syndromes. Mutations in TUBB2B have been reported in four patients and in one fetus with complex brain dysgenesis with asymmetrical, anteriorly predominant polymicrogyria.5
We performed TUBB2B mutation analysis in 128 patients in whom brain MRI had detected a spectrum of malformations of cortical development including polymicrogyria or pachygyria, to investigate how common TUBB2B mutations are in this spectrum of malformations and whether previously suggested imaging criteria need to be expanded.
Patients and methods
Patients
We selected 128 consecutive patients (61 females and 67 males) with a cortical pattern that was either clearly suggestive of polymicrogyria or pachygyria. Mutation analysis of the GPR56, TUBA1A, SRPX2 or LIS1, and DCX genes was performed in patients according to their malformation phenotype (see Supplementary Table S1).
Approval for the study was obtained from the Institutional Review Board of the Meyer Children's Hospital (IRB 00007816 AOU Meyer IRB #2). Clinical information and DNA samples were obtained after written informed consent from the patients’ parents.
TUBB2B mutation analysis
We screened the TUBB2B-coding region (GenBank Accession Number: NM_178012.4) in all 128 patients. DNA was extracted from peripheral blood using an automated DNA isolation robot (QIASymphony, QIAGEN, Hilden, Germany). The four coding exons of TUBB2B and their respective intron-exon boundaries were amplified by PCR (see Supplementary Table S2 for primer sequences and PCR conditions). PCR products were cycle sequenced on both strands using the BigDye Terminator v 1.1 chemistry (Applied Biosystems, Foster City, CA, USA) and run on a 3130XL genetic analyzer (Applied Biosystems).
The identified mutations were not found in an ethnic-matched cohort of 190 controls (380 alleles). TUBB2B-coding region polymorphisms identified in our patients and those included in the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/) are reported as online-only material (see Supplementary Table S3).
Protein structural modelling
The model of α-tubulin/β-tubulin dimer was built using Sus scrofa β-tubulin structure (PDB code 1TUB),6 which is identical to the human TUBB2B protein. The images in Figure 2 were rendered using UCSF Chimera (http://plato.cgl.ucsf.edu/chimera/).

Cartoon representation of the human α-tubulin/β-tubulin dimer. In blue, the mutations identified in the present study; in green, the mutations identified by Jaglin et al.5 The Asn256 residue, located in the β-tubulin H8 helix, is involved in the formation of the longitudinal contacts between α and β-tubulin at both the intradimer and interdimer interface. The Asp417 residue is located in the β-tubulin H12 helix, which represents a major site for interaction with motor proteins. The H12 helix contributes to generate lateral contacts between tubulin protofilaments. The H3 helix too, in which the Leu117 residue is located, is involved in the interaction between protofilaments. The Leu117Pro mutation substitutes a leucine with a proline that may act as a structural disruptor in the middle of regular secondary structure elements, such as α-helices.
Results
We identified missense mutations of TUBB2B in three unrelated patients.
Clinical and neuroimaging findings
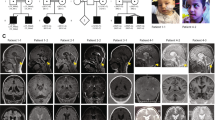
All three patients were born from unrelated healthy parents. Patient 1 was an 8-year-old girl. Development was delayed with sitting at 8 months and autonomous walking at 17 months. At age 2 years and 6 months, she exhibited microcephaly, oromotor dyspraxia, sialorrhea and mild hypotonia. Language was severely delayed. Generalized sleep-related tonic seizures started at age 11 months and atypical absences at age 3. Interictal EEG showed electrical status epilepticus during sleep (ESES). Brain MRI revealed diffuse polymicrogyria, more severe in the perisylvian regions (Figures 1a–d) and a small pons. Corpus callosum and cerebellum were normal.

Brain MRI scans of the three individuals with TUBB2B mutations. (a–d): Patient 1; (e–h): patient 2; (i–l): patient 3. (a, e, i): T1-weighed sagittal sections passing through the midline show a small pons in patient 1 (a), a thin corpus callosum in patient 2 (e), and normal structures in patient 3 (i). (b, j) are T1-weighed sagittal sections passing through the sylvian cortex; (f) is a proton density (PD)-weighed axial section passing through the semiovale centers. Note the thickening of the perisylvian cortex, with abnormal orientation of the sylvian fissure in patient 1 (b) and 3 (j), and pachygyria of the post-rolandic cortex in patient 2 (f). (c, g, k) are axial T1- (c), PD (g), and T2-weighed (k) sections. In patient 1 (c), the gyral pattern is diffusely abnormal whereas there is prominent thickening in the perisylvian cortex. In patient 2 (g), there is pronounced thickening in the posterior perisylvian cortex, with abnormal folding, which is more prominent on the left. In patient 3 (k), the cortical mantle is diffusely abnormal, with prominent infoldings in the posterior aspect of the abnormally oriented sylvian fissures, more on the left (black arrowhead). This aspect is consistent with both polymicrogyria and cobblestone-type cortex. (d, h, l) are coronal T1 (d), T2 (h), and PD-weighed (l) cuts through the posterior sylvian fissures. The gyral pattern is diffusely abnormal, with prominent thickening and infolding of the sylvian fissures.
Patient 2 was a 2-year-old boy. Brain MRI, performed at 6 months, revealed pachygyria with cortical thickening and irregular folding in the post-rolandic and parieto-posterior temporal regions (Figures 1e–h). The corpus callosum was thin and the cerebellum appeared to be normal. At the age of 1 year, he exhibited microcephaly, postural delay, mild hypotonia and sialorrhea. EEG was unremarkable. At the age of 2 years, he was nonverbal and exhibited psychomotor skills at the level of a 15-month-old.
Patient 3 was a 14-year-old boy. Autonomous walking was reached at 2 years. At the age of 3 years, speech was limited to a few isolated words. At the age of 10 years, neurological examination revealed microcephaly, moderate mental retardation, oromotor dyspraxia and sialorrhea. EEG was unremarkable. Brain MRI revealed diffuse polymicrogyria (Figures 1i–l) with a remarkably irregular cortical surface, reminiscent of the cobblestone cortex. There was a minor cavum septi pellucidi. The corpus callosum and cerebellum were normal.
TUBB2B mutation analysis
Patient 1 harbored a c.1249G>A substitution leading to a p.Asp417Asn mutation in the C-terminal domain of the protein; patient 2 harbored a c.767A>G substitution leading to a p.Asn256Ser mutation in the intermediate domain; and patient 3 had a c.350T>C substitution leading to a p.Leu117Pro mutation in the N-terminal domain. The mutations identified in patients 1 and 2 were de novo. For patient 3, the only available parent for testing was the mother, who was mutation-negative.
Protein structural modeling
We examined the potential effects of the mutated residues on the known structure of the β-tubulin polypeptide.6 The Asn256Ser substitution is located in the H8 helix of the intermediate domain, the Asp417Asn substitution falls in the H12 helix of the C-terminal domain and the Leu117Pro change in the H3 helix of the N-terminal domain (Figure 2).
Discussion
Tubulins have an important role in cortical malformations.7 Mutations of TUBA1A have been consistently associated with lissencephaly8 and, more rarely, with polymicrogyria.9 Mutations of TUBA8 have been associated with recessive polymicrogyria with optic nerve hypoplasia,3 and mutations of TUBB3 cause frontal polymicrogyria or simplified and disorganized gyral patterning.10 De novo TUBB2B mutations, all missense, have been identified in four patients and in one fetus with bilateral, asymmetrical and anteriorly predominant polymicrogyria.5 Neuropathological studies in fetuses with TUBA1A and TUBB2B mutations suggest that tubulin-related cortical dysgeneses are neuronal migration disorders with mislocation of developing neurons within the cortex.7 Functional studies suggest that abnormal neuronal migration would result from defective interactions in the tubulin heterodimer assembly and altered three-dimensional conformation of the tubulins, with compromised interaction with microtubule-associated proteins or microtubule motors.11
This study brings to eight the number of individuals with TUBB2B mutations reported so far. We identified three new TUBB2B mutations in as many unrelated patients with rather symmetrical malformations of cortical development, including polymicrogyria in two patients, one having a cobblestone-like cortex, and bilateral regional pachygyria in one. All mutations involved residues that are highly conserved across a wide range of vertebrate tubulins (data not shown). The Asn256Ser mutation affects a residue involved in the formation of the longitudinal contacts between α-tubulin and β-tubulin, possibly impairing the α-tubulin/β-tubulin heterodimer formation. This alteration could also lead to microtubule instability. The Asp417Asn mutation may alter the interactions with either motor proteins or between protofilaments. The Leu117Pro mutation probably disrupts the H3 helix, impairing the lateral contacts between protofilaments (Figure 2). Overall, the three mutations we identified may alter the proper functions of microtubules. Functional studies will be required to unravel the effects of these TUBB2B mutations on microtubule assembly.
Polymicrogyria is associated with a wide array of patterns and syndromes and with mutations in several known genes, although additional causative genes are likely to exist. The pathogenesis of polymicrogyria is poorly understood. Bilateral perisylvian polymicrogyria (BPP) is usually symmetrical and genetically heterogeneous.12 Mutations of SRPX2 have been related to BPP, but the role of this gene needs to be confirmed.2 Asymmetric BPP, with a striking predisposition for the right hemisphere, has been associated with 22q11.2 deletions.12 Mutations of GPR56 have been reported in patients with bilateral frontoparietal polymicrogyria,4 a recessive disorder with MRI characteristics resembling those of the cobblestone malformations spectrum.2 TUBB2B mutations have previously been associated with anteriorly predominant asymmetrical polymicrogyria,5 more severe in the fronto-parietal areas, and involving the perisylvian region with left-sided predominance. Jaglin et al5 demonstrated that TUBB2B mutations can lead to overmigration of MAP2-positive neurons through breaches in the pial basement membrane, leading to cobblestone-like structural changes. Basal ganglia appeared dysmorphic in previously reported patients, with fusion of the caudate and putamen nuclei, hypoplasia of the internal capsule and corpus callosum agenesis. The cortical malformation was rather symmetrical in our patients; we only observed a thin corpus callosum, as well as a hypoplastic internal capsulae in patient 2 and cavum septi pellucidi in patient 3.
We found TUBB2B mutations in about 2.3% of patients with malformations of cortical development we had selected, assuming mutations of this gene might cause a wider malformation spectrum than described in the only available report. In the group we screened, additional patients who also exhibited anatomic features similar to those of patients carrying TUBB2B mutations were mutation-negative. Criteria for searching TUBB2B mutations5 are still unclear. Other members of the tubulin family, that number >20 proteins, might be involved in causing a wide range of malformations of cortical development.
References
Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB : A developmental and genetic classification for malformations of cortical development. Neurology 2005; 65: 1873–1887.
Guerrini R, Dobyns WB, Barkovich AJ : Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. TrendsNeurosci 2008; 31: 154–162.
Abdollahi MR, Morrison E, Sirey T et al: Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am J Hum Genet 2009; 85: 737–744.
Piao X, Hill RS, Bodell A et al: G protein-coupled receptor-dependent development of human frontal cortex. Science 2004; 303: 2033–2036.
Jaglin XH, Poirier K, Saillour Y et al: Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet 2009; 41: 746–752.
Nogales E, Wolf SG, Downing KH : Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998; 391: 199–203.
Jaglin XH, Chelly J : Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet 2009; 25: 555–566.
Kumar RA, Pilz DT, Babatz TD et al: TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet 2010; 19: 2817–2827.
Jansen AC, Oostra A, Desprechins B et al: TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology 2011; 76: 988–992.
Poirier K, Saillour Y, Bahi-Buisson N et al: Mutations in the neuronal β-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet 2010; 19: 4462–4473.
Friocourt G, Marcorelles P, Saugier-Veber P, Quille ML, Marret S, Laquerrière A : Role of cytoskeletal abnormalities in the neuropathology and pathophysiology of type I lissencephaly. Acta Neuropathol 2011; 121: 149–170.
Guerreiro MM, Hage SR, Guimarães CA et al: Developmental language disorder associated with polymicrogyria. Neurology 2002; 59: 245–250.
Acknowledgements
We thank patients and their families for their participation to the study. This research was supported by a grant from the EU Sixth Framework Thematic Priority Life Sciences, Genomics and Biotechnology for Health, contract number LSH-CT-2006-037315 (EPICURE) (to RG) and from the ERA-Net for Research Programmes on Rare Diseases (E-RARE-2), (TUB-GENCODEV) (to RG).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Guerrini, R., Mei, D., Cordelli, D. et al. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur J Hum Genet 20, 995–998 (2012). https://doi.org/10.1038/ejhg.2012.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.21
Keywords
This article is cited by
-
Novel maternal duplication of 6p22.3-p25.3 with subtelomeric 6p25.3 deletion: new clinical findings and genotype–phenotype correlations
Molecular Cytogenetics (2023)
-
Insights on the Role of α- and β-Tubulin Isotypes in Early Brain Development
Molecular Neurobiology (2023)
-
Cul3 regulates cytoskeleton protein homeostasis and cell migration during a critical window of brain development
Nature Communications (2021)
-
Associated analysis of PER1/TUBB2B with endometrial cancer development caused by circadian rhythm disorders
Medical Oncology (2020)
-
Tubulinopathies continued: refining the phenotypic spectrum associated with variants in TUBG1
European Journal of Human Genetics (2018)
