
Unravelling the Clinical Co-Morbidity and Risk Factors Associated with Agenesis of the Corpus Callosum

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Data Source and Study Design
2.2. Data Collation and Analysis
2.3. Statistical Analysis
3. Results
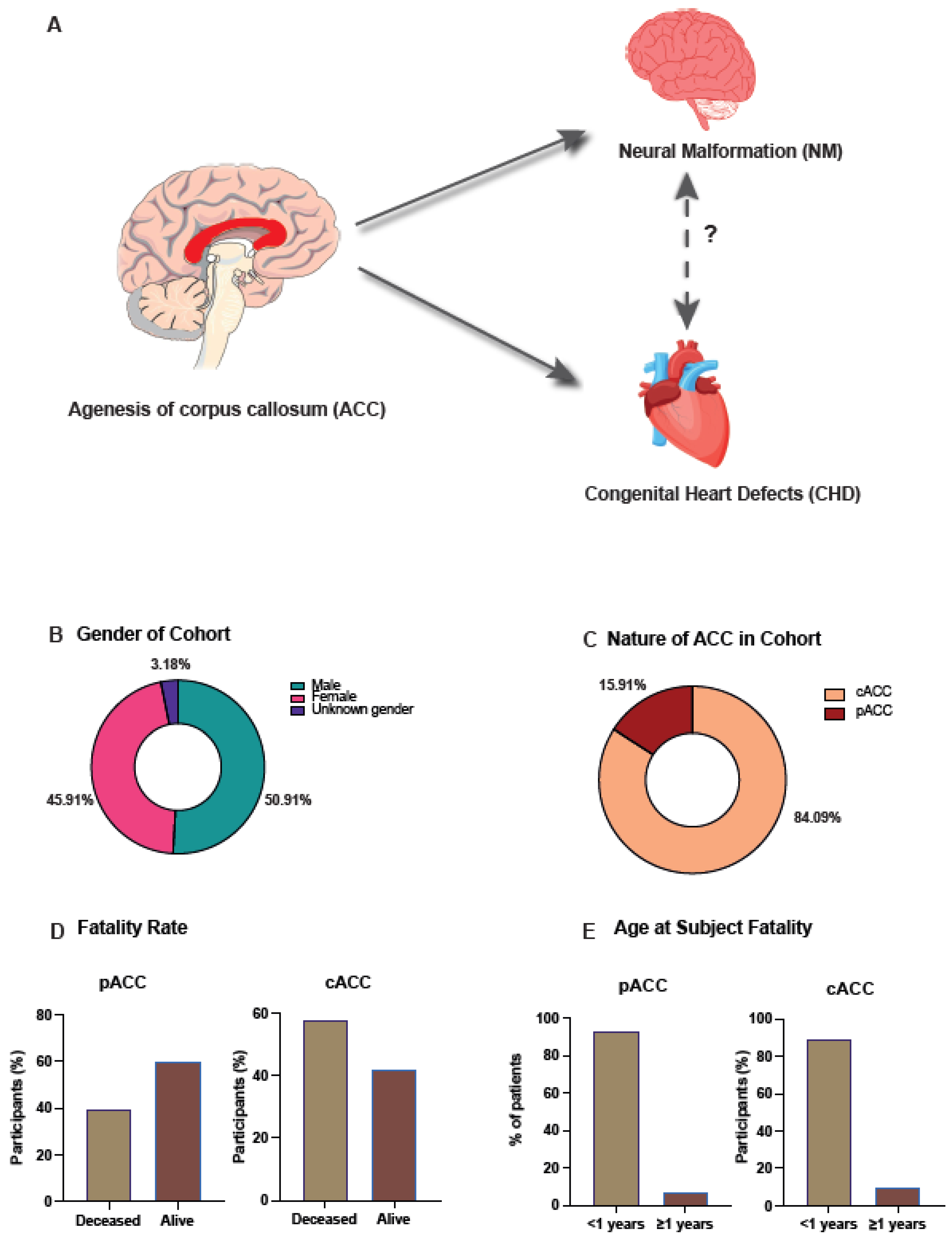
3.1. Epidemiological Analysis of ACC in All Wales Cohort
3.2. Complete Agenesis of the Corpus Callosum Is the Most Observed Morphology
3.3. Ventriculomegaly/Hydrocephalus Was the Most Observed Neural Malformation in Those with ACC
3.4. Ventricular Septal Defect Was the Most Observed Congenital Heart Disorder within the Cohort
3.5. Association between NM and CHD within ACC Cohort Was Not Significant
3.6. Risk Factors Analysis That Can Influence the Aetiology of ACC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schell-Apacik, C.C.; Wagner, K.; Bihler, M.; Ertl-Wagner, B.; Heinrich, U.; Klopocki, E.; Kalscheuer, V.M.; Muenke, M.; von Voss, H. Agenesis and dysgenesis of the corpus callosum: Clinical, genetic and neuroimaging findings in a series of 41 patients. Am. J. Med. Genet. A 2008, 146A, 2501–2511. [Google Scholar] [CrossRef]
- Hofman, J.; Hutny, M.; Sztuba, K.; Paprocka, J. Corpus callosum agenesis: An insight into the etiology and spectrum of symptoms. Brain Sci. 2020, 10, 625. [Google Scholar] [CrossRef]
- Tomasch, J. Size, distribution, and number of fibres in the human Corpus Callosum. Anat. Rec. 1954, 119, 119–135. [Google Scholar] [CrossRef]
- Kamnasaran, D. Corpus Callosum: Agenesis. In Encyclopedia of Neuroscience; Squire, L., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 163–173. [Google Scholar]
- Achiron, R.; Achiron, A. Development of the human fetal corpus callosum: A high-resolution, cross-sectional sonographic study. Ultrasound Obstet. Gynecol. 2001, 18, 343–347. [Google Scholar] [CrossRef]
- Rakic, P.; Yakovlev, P.I. Development of the corpus callosum and cavum septi in man. J. Comp. Neurol. 1968, 132, 45–72. [Google Scholar] [CrossRef]
- Goldstein, A.; Covington, B.; Mahabadi, N.; Mesfin, F. Neuroanatomy, Corpus Callosum. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Shahid, S.; Mytilinaios, D. Corpus Callosum. 2022. Available online: https://www.kenhub.com/en/library/anatomy/corpus-callosum (accessed on 30 January 2022).
- Paul, L.K.; Brown, W.S.; Adolphs, R.; Tyszka, J.M.; Richards, L.J.; Mukherjee, P.; Sherr, E.H. Agenesis of the corpus callosum: Genetic, developmental and functional aspects of connectivity. Nat. Rev. Neurosci. 2007, 8, 287–299. [Google Scholar] [CrossRef]
- Romaniello, R.; Marelli, S.; Giorda, R.; Bedeschi, M.F.; Bonaglia, M.C.; Arrigoni, F.; Triulzi, F.; Bassi, M.T.; Borgatti, R. Clinical Characterization, Genetics, and Long-Term Follow-up of a Large Cohort of Patients With Agenesis of the Corpus Callosum. J. Child Neurol. 2017, 32, 60–71. [Google Scholar] [CrossRef]
- Kim, Y.U.; Park, E.S.; Jung, S.; Suh, M.; Choi, H.S.; Rha, D.-W. Clinical features and associated abnormalities in children and adolescents with corpus callosal anomalies. Ann. Rehabil. Med. 2014, 38, 138–143. [Google Scholar] [CrossRef]
- Bedeschi, M.F.; Bonaglia, M.C.; Grasso, R.; Pellegri, A.; Garghentino, R.R.; Battaglia, M.A.; Panarisi, A.M.; Di Rocco, M.; Balottin, U.; Bresolin, N.; et al. Agenesis of the Corpus Callosum: Clinical and Genetic Study in 63 Young Patients. Pediatr. Neurol. 2006, 34, 186–193. [Google Scholar] [CrossRef]
- Guadarrama-Ortiz, P.; Choreño-Parra, J.A.; de la Rosa-Arredondo, T. Isolated agenesis of the corpus callosum and normal general intelligence development during postnatal life: A case report and review of the literature. J. Med. Case Rep. 2020, 14, 28. [Google Scholar] [CrossRef]
- Glass, H.C.; Shaw, G.M.; Ma, C.; Sherr, E.H. Agenesis of the corpus callosum in California 1983-2003: A population-based study. Am. J. Med. Genet. A 2008, 146A, 2495–2500. [Google Scholar] [CrossRef]
- Dowden, L.; Tucker, D.; Morgan, S.; Uzun, O.; Syed, Y.A. Contribution of Congenital Heart Disorders Associated With Copy Number Variants in Mediating Risk for Brain Developmental Disorders: Evidence From 20-Year Retrospective Cohort Study. Front. Cardiovasc. Med. 2021, 8, 655463. [Google Scholar] [CrossRef]
- Sun, L.; Macgowan, C.K.; Sled, J.G.; Yoo, S.-J.; Manlhiot, C.; Porayette, P.; Grosse-Wortmann, L.; Jaeggi, E.; McCrindle, B.W.; Kingdom, J.; et al. Reduced Fetal Cerebral Oxygen Consumption Is Associated With Smaller Brain Size in Fetuses With Congenital Heart Disease. Circulation 2015, 131, 1313–1323. [Google Scholar] [CrossRef]
- Morton, P.D.; Ishibashi, N.; Jonas, R.A. Neurodevelopmental Abnormalities and Congenital Heart Disease: Insights into Altered Brain Maturation. Circ. Res. 2017, 120, 960–977. [Google Scholar] [CrossRef]
- Mattson, S.N.; Riley, E.P.; Gramling, L.; Delis, D.C.; Jones, K.L. Neuropsychological Comparison of Alcohol-Exposed Children With or Without Physical Features of Fetal Alcohol Syndrome. Neuropsychology 1998, 12, 146–153. [Google Scholar] [CrossRef]
- Welsh Government. Welsh Index of Multiple Deprivation. 2019. Available online: https://gov.wales/welsh-index-multiple-deprivation-full-index-update-ranks-2019 (accessed on 18 April 2022).
- European Commission. European Platfrom on Rare Disease Registration. 2022. Available online: https://eu-rd-platform.jrc.ec.europa.eu/ (accessed on 10 June 2022).
- GraphPad Software. GraphPad Prism, 9.3.1; GraphPad Software: San Diego, CA, USA, 2022.
- Microsoft Corporation. Microsoft Excel, 16.60; Microsoft Corporation: Redmond, WA, USA, 2022.
- Taylor, M.; David, A.S. Agenesis of the corpus callosum: A United Kingdom series of 56 cases. J. Neurol. Neurosurg. Psychiatry 1998, 64, 131–134. [Google Scholar] [CrossRef]
- Goodyear, P.W.A.; Bannister, C.M.; Russell, S.; Rimmer, S. Outcome in Prenatally Diagnosed Fetal Agenesis of the Corpus callosum. Fetal Diagn. Ther. 2001, 16, 139–145. [Google Scholar] [CrossRef]
- Wiechec, M.; Nocun, A.; Knafel, A.; Beithon, J.; Stettner, D. Four Steps in Diagnosing Complete Agenesis of the Corpus Callosum in Prenatal Life. Ultraschall Med. 2015, 37, 92–99. [Google Scholar] [CrossRef]
- Santo, S.; D’Antonio, F.; Homfray, T.; Rich, P.; Pilu, G.; Bhide, A.; Thilaganathan, B.; Papageorghiou, A.T. Counseling in fetal medicine: Agenesis of the corpus callosum. Ultrasound Obstet. Gynecol. 2012, 40, 513–521. [Google Scholar] [CrossRef]
- Ozyüncü, O.; Yazıcıoğlu, A.; Turğal, M. Antenatal diagnosis and outcome of agenesis of corpus callosum: A retrospective review of 33 cases. J. Turk. Ger. Gynecol. Assoc. 2014, 15, 18–21. [Google Scholar] [CrossRef]
- Ballardini, E.; Marino, P.; Maietti, E.; Astolfi, G.; Neville, A.J. Prevalence and associated factors for agenesis of corpus callosum in Emilia Romagna (1981–2015). Eur. J. Med. Genet. 2018, 61, 524–530. [Google Scholar] [CrossRef]
- Hetts, S.W.; Sherr, E.H.; Chao, S.; Gobuty, S.; Barkovich, A.J. Anomalies of the corpus callosum: An MR analysis of the phenotypic spectrum of associated malformations. Am. J. Roentgenol. 2006, 187, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, J.; Shao, X. Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990–2017. Medicine 2020, 99, e20593. [Google Scholar] [CrossRef] [PubMed]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, C.J.; Smith, Z.G.; Rasool, H.; Cullen, K.; Ghosh, M.; Woolley, T.E.; Uzun, O.; Loh, N.R.; Tucker, D.; Syed, Y.A. Unravelling the Clinical Co-Morbidity and Risk Factors Associated with Agenesis of the Corpus Callosum. J. Clin. Med. 2023, 12, 3623. https://doi.org/10.3390/jcm12113623
Smith CJ, Smith ZG, Rasool H, Cullen K, Ghosh M, Woolley TE, Uzun O, Loh NR, Tucker D, Syed YA. Unravelling the Clinical Co-Morbidity and Risk Factors Associated with Agenesis of the Corpus Callosum. Journal of Clinical Medicine. 2023; 12(11):3623. https://doi.org/10.3390/jcm12113623
Chicago/Turabian StyleSmith, Callum J., Zoey G. Smith, Hania Rasool, Katie Cullen, Meghana Ghosh, Thomas E. Woolley, Orhan Uzun, Ne Ron Loh, David Tucker, and Yasir Ahmed Syed. 2023. "Unravelling the Clinical Co-Morbidity and Risk Factors Associated with Agenesis of the Corpus Callosum" Journal of Clinical Medicine 12, no. 11: 3623. https://doi.org/10.3390/jcm12113623