

Identification of Compound Heterozygous EVC2 Gene Variants in Two Mexican Families with Ellis–van Creveld Syndrome
 , , and
, , and
Abstract
:1. Introduction
2. Cases and Methods
2.1. Case Report
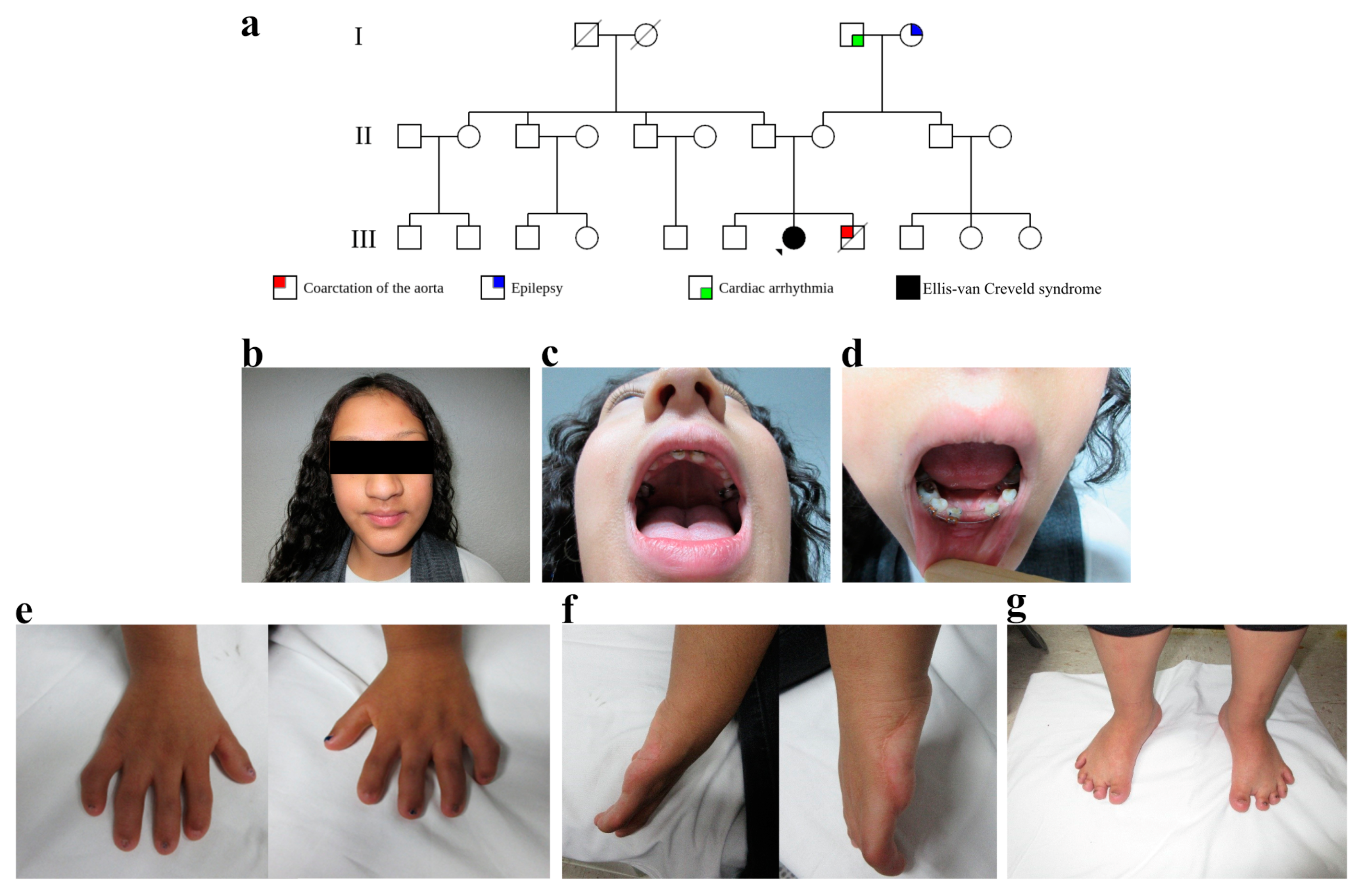
2.1.1. Case 1
2.1.2. Case 2
2.2. Molecular Genetic Studies
2.2.1. DNA Isolation
2.2.2. Whole Exome Sequencing
2.2.3. READ Mapping and Variant Analysis
2.2.4. PCR Amplification and Sanger Sequencing
- 5′AGAATGGCGTGAACCTAGGA3′ and 5′TCCACTGTGCACTAACGCTT3′ for exon 2;
- 5′CTGGTAAGCACACGGTACAT 3′ and 5′TTGAAAACTGTCAGGTACCCT 3′ for exon 4;
- 5′TGATAAATTCCCAGGCCCTC3′ and 5′CCACTGTGAGGATTAGGAGA3′ for exon 5;
- 5′GAGATTGTTGGGGAAAAGGC3′ and 5′GGCACTCACATGAAGATCAG3′ for exon 14.
2.2.5. 3D-Modeling
3. Results
4. Discussion
5. Conclusions
6. Web Sources
- HGMD: ”http://www.hgmd.cf.ac.uk” (accessed on 15 December 2022),
- ClinVar: “https://www.ncbi.nlm.nih.gov/clinvar/?term=evc2%5Bgene%5D&redir=gene” (accessed on 15 December 2022),
- LSDBs: “https://www.humanvariomeproject.org/” (accessed on 15 December 2022),
- NHLBI Exome Sequencing Project: “https://evs.gs.washington.edu/EVS/” (accessed on 15 December 2022),
- 1000 Genomes: “http://www.internationalgenome.org” (accessed on 15 December 2022),
- dbSNP: “https://www.ncbi.nlm.nih.gov/snp/” (accessed on 15 December 2022),
- AlignGVGD: “http://agvgd.hci.utah.edu/” (accessed on 15 December 2022),
- MAPP: “http://mendel.stanford.edu/SidowLab/downloads/MAPP” (accessed on 15 December 2022),
- SNAP: “https://rostlab.org/services/snap2web” (accessed on 15 December 2022),
- LOVD3: “https://databases.lovd.nl/shared/variants/0000710769#00007281” (accessed on 28 January 2023),
- VARSOME: “https://varsome.com/gene/hg38/evc2” (accessed on 15 December 2022).
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, J.E.; Gleeson, J.G. A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 2011, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef]
- Zhang, W.; Taylor, S.P.; Ennis, H.A.; Forlenza, K.N.; Duran, I.; Li, B.; Sanchez, J.A.O.; Nevarez, L.; Nickerson, D.A.; Bamshad, M.; et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat. 2018, 39, 152–166. [Google Scholar] [CrossRef]
- Hammarsjö, A.; Pettersson, M.; Chitayat, D.; Handa, A.; Anderlid, B.-M.; Bartocci, M.; Basel, D.; Batkovskyte, D.; Beleza-Meireles, A.; Conner, P.; et al. High diagnostic yield in skeletal ciliopathies using massively parallel genome sequencing, structural variant screening and RNA analyses. J. Hum. Genet. 2021, 66, 995–1008. [Google Scholar] [CrossRef]
- Tan, W.; Lin, A.; Keppler-Noreuil, K. Cranioectodermal Dysplasia Synonym: Sensenbrenner Syndrome. 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK154653/ (accessed on 22 December 2022).
- Handa, A.; Voss, U.; Hammarsjö, A.; Grigelioniene, G.; Nishimura, G. Skeletal ciliopathies: A pattern recognition approach. Jpn. J. Radiol. 2020, 38, 193–206. [Google Scholar] [CrossRef]
- Cheney, R.W. Ellis-van creveld syndrome. Salem Press Encycl Health, Salem Press: Amenia, NY, USA. 2022, 2, 1–4. [Google Scholar]
- Ruiz-Perez, V.L.; Goodship, J.A. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to Hedgehog ligands. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.W.B.; Van Creveld, S. A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: Report of three cases. Arch. Dis. Child. 1940, 15, 65–84. [Google Scholar] [CrossRef] [Green Version]
- Baujat, G.; Le Merrer, M. Ellis-van creveld syndrome. Orphanet J. Rare Dis. 2007, 2, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, E.K.; Louie, K.; Kulkarni, A.; Yatabe, M.; Ruellas, A.C.D.O.; Snider, T.N.; Mochida, Y.; Cevidanes, L.H.S.; Mishina, Y.; Zhang, H. The Role of Ellis-Van Creveld 2(EVC2) in Mice During Cranial Bone Development. Anat. Rec. 2018, 301, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badano, J.L.; Mitsuma, N.; Beales, P.L.; Katsanis, N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genom. Hum. Genet. 2006, 7, 125–148. [Google Scholar] [CrossRef] [Green Version]
- Lauritano, D.; Attuati, S.; Besana, M.; Rodilosso, G.; Quinzi, V.; Marzo, G.; Carinci, F. Oral and craniofacial manifestations of Ellis-Van Creveld syndrome: A systematic review. Eur. J. Paediatr. Dent. 2019, 20, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, V.L.; Ide, S.E.; Strom, T.M.; Lorenz, B.; Wilson, D.; Woods, K.; King, L.; Francomano, C.; Freisinger, P.; Spranger, S.; et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000, 24, 283–286. [Google Scholar] [CrossRef]
- Huber, C.; Cormier-Daire, V. Ciliary disorder of the skeleton. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160, 165–174. [Google Scholar] [CrossRef]
- Shetty, D.C.; Singh, H.P.; Kumar, P.; Verma, C. Report of Two Siblings with Overlapping Features of Ellis-van Creveld and Weyers Acrodental Dysostosis. J. Clin. Imaging Sci. 2012, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Hampl, M.; Cela, P.; Szabo-Rogers, H.; Bosakova, M.K.; Dosedelova, H.; Krejci, P.; Buchtova, M. Role of Primary Cilia in Odontogenesis. J. Dent. Res. 2017, 96, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Tompson, S.; Ruiz-Perez, V.L.; Blair, H.J.; Barton, S.; Navarro, V.; Robson, J.L.; Wright, M.J.; Goodship, J.A. Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis-van Creveld syndrome patients. Hum. Genet. 2007, 120, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Aubert-Mucca, M.; Huber, C.; Baujat, G.; Michot, C.; Zarhrate, M.; Bras, M.; Boutaud, L.; Malan, V.; Attie-Bitach, T.; Clinical Contributors; et al. Ellis-Van Creveld Syndrome: Clinical and Molecular Analysis of 50 Individuals Diagnostics. J. Med. Genet. 2022, 60, 337–345. [Google Scholar] [CrossRef]
- Blair, H.J.; Tompson, S.; Liu, Y.-N.; Campbell, J.; MacArthur, K.; Ponting, C.P.; Ruiz-Perez, V.L.; A Goodship, J. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol. 2011, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Louie, K.W.; Mishina, Y.; Zhang, H. Molecular and cellular pathogenesis of ellis-van creveld syndrome: Lessons from targeted and natural mutations in animal models. J. Dev. Biol. 2020, 8, 25. [Google Scholar] [CrossRef]
- Thomas, D.C.; Moorthy, J.D.; Prabhakar, V.; Ajayakumar, A.; Pitchumani, P.K. Role of primary cilia and Hedgehog signaling in craniofacial features of Ellis–van Creveld syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Abramyan, J. Hedgehog signaling and embryonic craniofacial disorders. J. Dev. Biol. 2019, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 418–431. [Google Scholar] [CrossRef]
- Dworkin, S.; Boglev, Y.; Owens, H.; Goldie, S.J. The role of Sonic hedgehog in craniofacial patterning, morphogenesis and cranial neural crest survival. J. Dev. Biol. 2016, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, A.; Shalehin, N.; Takebe, H.; Shimo, T.; Irie, K. Sonic Hedgehog Signaling and Tooth Development. Int. J. Mol. Sci. 2020, 21, 1587. [Google Scholar] [CrossRef] [Green Version]
- Xavier, G.M.; Seppala, M.; Barrell, W.; Birjandi, A.A.; Geoghegan, F.; Cobourne, M.T. Hedgehog receptor function during craniofacial development. Dev. Biol. 2016, 415, 198–215. [Google Scholar] [CrossRef] [Green Version]
- Ginns, E.I.; Galdzicka, M.; Elston, R.C.; Song, Y.E.; Paul, S.M.; Egeland, J.A. Disruption of sonic hedgehog signaling in Ellis-van Creveld dwarfism confers protection against bipolar affective disorder. Mol. Psychiatry 2015, 20, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Caparrós-Martín, J.A.; Valencia, M.; Reytor, E.; Pacheco, M.; Fernandez, M.; Perez-Aytes, A.; Gean, E.; Lapunzina, P.; Peters, H.; Goodship, J.A.; et al. The ciliary EVC/EVC2 complex interacts with smo and controls hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. 2013, 22, 124–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Carvallo, P. Conceptos Sobre Genética Humana Para La Comprensión E Interpretación De Las Mutaciones En Cáncer Y Otras Patologías Hereditarias. Rev. Méd. Clín. Condes 2017, 28, 531–537. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Rubio, S.; Pacheco-Orozco, R.A.; Gómez, A.M.; Perdomo, S.; García-Robles, R. Secuenciación de nueva generación (NGS) de ADN: Presente y futuro en la práctica clínica. Univ. Med. 2020, 61, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Strom, S.P.; Lee, H.; Das, K.; Vilain, E.; Nelson, S.F.; Grody, W.W.; Deignan, J.L. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet. Med. 2014, 16, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. ProMod3—A versatile homology modelling toolbox. PLoS Comput. Biol. 2021, 17, e1008667. [Google Scholar] [CrossRef]
- Biasini, M.; Schmidt, T.; Bienert, S.; Mariani, V.; Studer, G.; Haas, J.; Johner, N.; Schenk, A.D.; Philippsen, A.; Schwede, T. OpenStructure: An integrated software framework for computational structural biology. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Kamiya, N.; Tsuji, T.; Takeda, H.; Scott, G.; Rajderkar, S.; Ray, M.K.; Mochida, Y.; Allen, B.; Lefebvre, V.; et al. Elevated Fibroblast Growth Factor Signaling Is Critical for the Pathogenesis of the Dwarfism in Evc2/Limbin Mutant Mice. PLoS Genet. 2016, 12, e1006510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D'Asdia, M.C.; Torrente, I.; Consoli, F.; Ferese, R.; Magliozzi, M.; Bernardini, L.; Guida, V.; Digilio, M.C.; Marino, B.; Dallapiccola, B.; et al. Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur. J. Med. Genet. 2013, 56, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Valencia, M.; Lapunzina, P.; Lim, D.; Zannolli, R.; Bartholdi, D.; Wollnik, B.; Al-Ajlouni, O.; Eid, S.S.; Cox, H.; Buoni, S.; et al. Widening the mutation spectrum of EVC and EVC2: Ectopic expression of Weyer variants in NIH 3T3 fibroblasts disrupts Hedgehog signaling. Hum Mutat. 2009, 30, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, V.L.; Tompson, S.; Blair, J.H.; Espinoza-Valdez, C.; Lapunzina, P.; Silva, E.O.; Hamel, B.; Gibbs, J.L.; Young, I.D.; Wright, M.J.; et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am. J. Hum. Genet. 2003, 72, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Chivers, M.; Královičová, J.; Romano, M.; Baralle, M.; Krainer, A.; Vořechovský, I. Aberrant 5′ splice sites in human disease genes: Mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007, 35, 4250–4263. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Classified Variants (UniProt, ClinVar, VarSome & PubMed) | ||||||
|---|---|---|---|---|---|---|
| n = 1021 | ||||||
| Pathogenic | Uncertain Significance | Benign | ||||
| n = 224 | n = 250 | n = 547 | ||||
| Coding Impact | Pathogenic | Likely Pathogenic | Uncertain Significance (VUS) | Likely Benign | Benign | Total |
| Synonymous | 0 | 0 | 29 | 389 | 24 | 442 |
| Missense | 5 | 2 | 188 | 10 | 27 | 232 |
| Nonsense | 63 | 18 | 2 | 0 | 0 | 83 |
| Frameshift | 50 | 41 | 3 | 0 | 0 | 94 |
| Inframe Indel | 0 | 1 | 13 | 0 | 0 | 14 |
| Splice junction loss | 7 | 37 | 0 | 0 | 0 | 44 |
| Non-coding | 0 | 0 | 15 | 82 | 15 | 112 |
| Total | 125 | 99 | 250 | 481 | 66 | 1021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negrete-Torres, N.; Chima-Galán, M.d.C.; Sierra-López, E.A.; Sánchez-Ramos, J.; Álvarez-González, I.; Reyes-Reali, J.; Mendoza-Ramos, M.I.; Garrido-Guerrero, E.; Amato, D.; Méndez-Catalá, C.F.; et al. Identification of Compound Heterozygous EVC2 Gene Variants in Two Mexican Families with Ellis–van Creveld Syndrome. Genes 2023, 14, 887. https://doi.org/10.3390/genes14040887
Negrete-Torres N, Chima-Galán MdC, Sierra-López EA, Sánchez-Ramos J, Álvarez-González I, Reyes-Reali J, Mendoza-Ramos MI, Garrido-Guerrero E, Amato D, Méndez-Catalá CF, et al. Identification of Compound Heterozygous EVC2 Gene Variants in Two Mexican Families with Ellis–van Creveld Syndrome. Genes. 2023; 14(4):887. https://doi.org/10.3390/genes14040887
Chicago/Turabian StyleNegrete-Torres, Nancy, María del Carmen Chima-Galán, Ernesto Antonio Sierra-López, Janet Sánchez-Ramos, Isela Álvarez-González, Julia Reyes-Reali, María Isabel Mendoza-Ramos, Efraín Garrido-Guerrero, Dante Amato, Claudia Fabiola Méndez-Catalá, and et al. 2023. "Identification of Compound Heterozygous EVC2 Gene Variants in Two Mexican Families with Ellis–van Creveld Syndrome" Genes 14, no. 4: 887. https://doi.org/10.3390/genes14040887