
Dental Anomalies in Ciliopathies: Lessons from Patients with BBS2, BBS7, and EVC2 Mutations
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
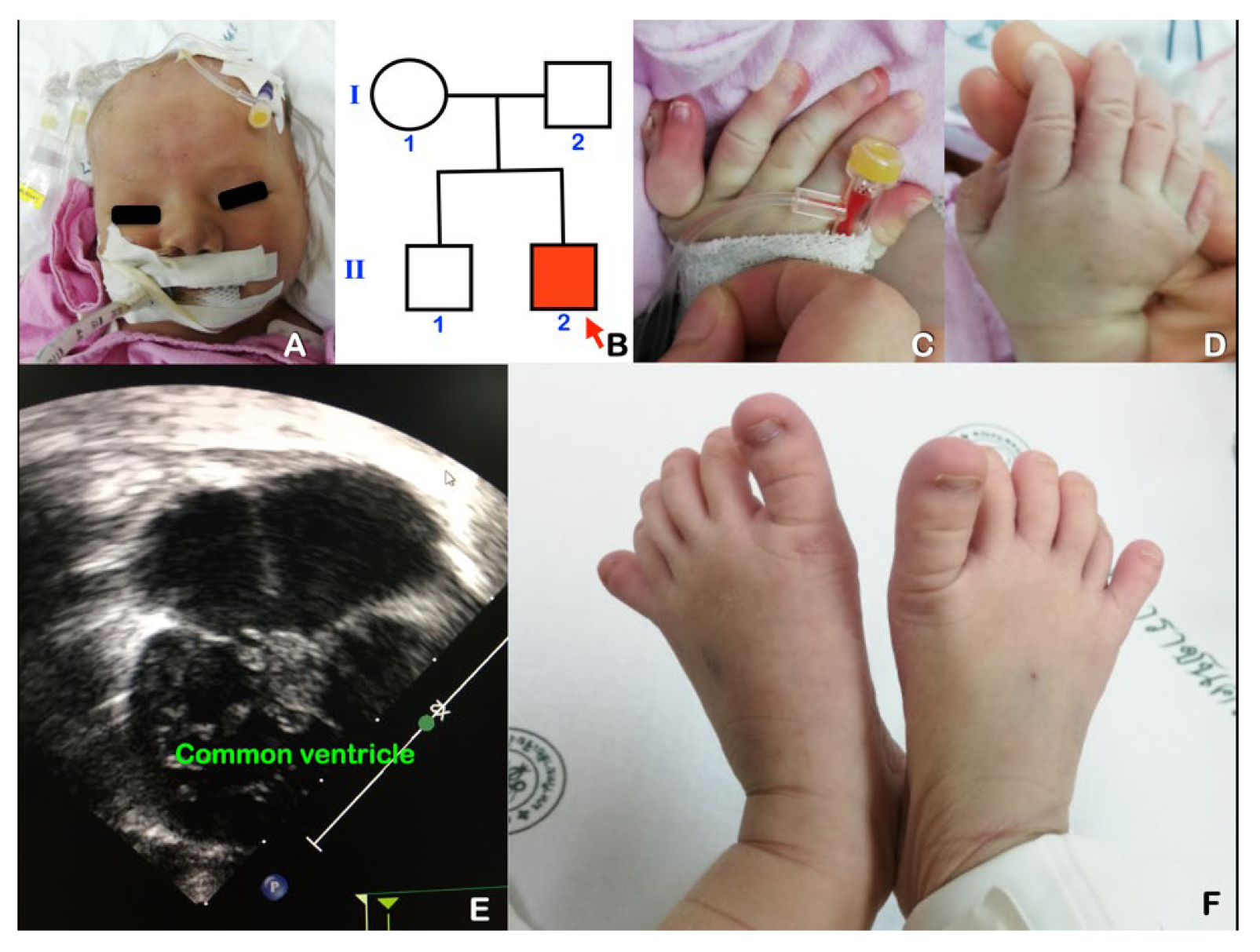
Abstract
:1. Introduction
2. Patients and Methods
2.1. Subjects
2.2. Whole Exome Sequencing and Mutation Analysis
3. Results
Whole Exome Sequencing and Mutation Analysis
4. Discussion
4.1. BBS2 and BBS7 Mutations and Impaired BBSome Complex
4.2. Oral Manifestations in EVC and BBS
4.3. EVC, BBS, and Abnormal Hh and Wnt Signaling
4.4. Ciliopathy, WNT Signaling, and Dental Anomalies

5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.S.; Chuang, J.Z.; Sung, C.H. The Biology of Ciliary Dynamics. Cold Spring Harb. Perspect. Biol. 2017, 9, a027904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priya, S.; Nampoothiri, S.; Sen, P.; Sripriya, S. Bardet-Biedl syndrome: Genetics, molecular pathophysiology, and disease management. Indian J. Ophthalmol. 2016, 64, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, Y.I.; Lin, C.; Chuang, P.T. Hedgehog signaling from the primary cilium to the nucleus: An emerging picture of ciliary localization, trafficking and transduction. Curr. Opin. Genet. Dev. 2013, 23, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell’s antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef]
- Lee, J.E.; Gleeson, J.G. A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 2011, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Borgström, M.K.; Riise, R.; Tornqvist, K.; Granath, L. Anomalies in the permanent dentition and other oral findings in 29 individuals with Laurence-Moon-Bardet-Biedl syndrome. J. Oral. Pathol. Med. 1996, 25, 86–89. [Google Scholar] [CrossRef]
- Andersson, E.M.; Axelsson, S.; Gjølstad, L.F.; Storhaug, K. Taurodontism: A minor diagnostic criterion in Laurence-Moon/Bardet-Biedl syndromes. Acta Odontol. Scand. 2013, 71, 1671–1674. [Google Scholar] [CrossRef]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Hampl, M.; Cela, P.; Szabo-Rogers, H.L.; Kunova Bosakova, M.; Dosedelova, H.; Krejci, P.; Buchtova, M. Role of Primary Cilia in Odontogenesis. J. Dent. Res. 2017, 96, 965–974. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef] [Green Version]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peränen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Nishimura, D.; Vogel, T.; Shao, J.; Swiderski, R.; Yin, T.; Searby, C.; Carter, C.S.; Kim, G.; Bugge, K.; et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J. Cell Sci. 2013, 126, 2372–2380. [Google Scholar] [CrossRef] [Green Version]
- Badano, J.L.; Katsanis, N. Beyond Mendel: An evolving view of human genetic disease transmission. Nat. Rev. Genet. 2002, 3, 779–789. [Google Scholar] [CrossRef]
- Klink, B.U.; Gatsogiannis, C.; Hofnagel, O.; Wittinghofer, A.; Raunser, S. Structure of the human BBSome core complex. Elife 2020, 9, e53910. [Google Scholar] [CrossRef]
- Blacque, O.E.; Reardon, M.J.; Li, C.; McCarthy, J.; Mahjoub, M.R.; Ansley, S.J.; Badano, J.L.; Mah, A.K.; Beales, P.L.; Davidson, W.S.; et al. Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 2004, 18, 1630–1642. [Google Scholar] [CrossRef]
- Zhang, Q.; Seo, S.; Bugge, K.; Stone, E.M.; Sheffield, V.C. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum. Mol. Genet. 2012, 21, 1945–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennekam, R.C.; Allanson, J.E.; Krantz, I.D. Gorlin’s Syndromes of the Head and Neck; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Corbit, K.C.; Shyer, A.E.; Dowdle, W.E.; Gaulden, J.; Singla, V.; Chen, M.H.; Chuang, P.T.; Reiter, J.F. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell. Biol. 2008, 10, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Balmer, S.; Dussert, A.; Collu, G.M.; Benitez, E.; Iomini, C.; Mlodzik, M. Components of Intraflagellar Transport Complex A Function Independently of the Cilium to Regulate Canonical Wnt Signaling in Drosophila. Dev. Cell 2015, 34, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Chen, S.; Cheng, D.; Jing, W.; Helms, J.A. Primary cilia integrate hedgehog and Wnt signaling during tooth development. J. Dent. Res. 2014, 93, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, J.M.; Liu, Y.; Zaghloul, N.A.; Leitch, C.C.; Lawson, S.S.; Kato, M.; Beachy, P.A.; Beales, P.L.; DeMartino, G.N.; Fisher, S.; et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007, 39, 1350–1360. [Google Scholar] [CrossRef]
- Zaghloul, N.A.; Katsanis, N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Investig. 2009, 119, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.J.; Tompson, S.; Liu, Y.N.; Campbell, J.; MacArthur, K.; Ponting, C.P.; Ruiz-Perez, V.L.; Goodship, J.A. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol. 2011, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Pusapati, G.V.; Hughes, C.E.; Dorn, K.V.; Zhang, D.; Sugianto, P.; Aravind, L.; Rohatgi, R. EFCAB7 and IQCE regulate hedgehog signaling by tethering the EVC-EVC2 complex to the base of primary cilia. Dev. Cell 2014, 28, 483–496. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Takeda, H.; Tsuji, T.; Kamiya, N.; Rajderkar, S.; Louie, K.; Collier, C.; Scott, G.; Ray, M.; Mochida, Y.; et al. Generation of Evc2/Limbin global and conditional KO mice and its roles during mineralized tissue formation. Genesis 2015, 53, 612–626. [Google Scholar] [CrossRef]
- Ruiz-Perez, V.L.; Blair, H.J.; Rodriguez-Andres, M.E.; Blanco, M.J.; Wilson, A.; Liu, Y.N.; Miles, C.; Peters, H.; Goodship, J.A. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 2007, 134, 2903–2912. [Google Scholar] [CrossRef] [Green Version]
- Nakatomi, M.; Hovorakova, M.; Gritli-Linde, A.; Blair, H.J.; MacArthur, K.; Peterka, M.; Lesot, H.; Peterkova, R.; Ruiz-Perez, V.L.; Goodship, J.A.; et al. Evc regulates a symmetrical response to Shh signaling in molar development. J. Dent. Res. 2013, 92, 222–228. [Google Scholar] [CrossRef]
- Zhang, H.; Takeda, H.; Tsuji, T.; Kamiya, N.; Kunieda, T.; Mochida, Y.; Mishina, Y. Loss of Function of Evc2 in Dental Mesenchyme Leads to Hypomorphic Enamel. J. Dent. Res. 2017, 96, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Han, D.; Zhang, J.; Zhao, H.; Feng, H. Two novel heterozygous mutations of EVC2 cause a mild phenotype of Ellis-van Creveld syndrome in a Chinese family. Am. J. Med. Genet. A 2011, 155, 2131–2136. [Google Scholar] [CrossRef]
- Pawlaczyk-Kamieńska, T.; Winiarska, H.; Kulczyk, T.; Cofta, S. Dental Anomalies in Rare, Genetic Ciliopathic Disorder-A Case Report and Review of Literature. Int. J. Environ. Res. Public Health 2020, 17, 4337. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Liu, M.; Zhang, S.; Wan, H.; Zhang, Q.; Yue, R.; Yan, X.; Wang, X.; Wang, Z.; Sun, Y. Essential Role of IFT140 in Promoting Dentinogenesis. J. Dent. Res. 2018, 97, 423–431. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Y.; Ma, C.; Li, B. MicroRNA-30a suppresses the proliferation, migration and invasion of human renal cell carcinoma cells by directly targeting ADAM9. Oncol. Lett. 2018, 16, 3038–3044. [Google Scholar] [CrossRef] [Green Version]
- Ohazama, A.; Haycraft, C.J.; Seppala, M.; Blackburn, J.; Ghafoor, S.; Cobourne, M.; Martinelli, D.C.; Fan, C.M.; Peterkova, R.; Lesot, H.; et al. Primary cilia regulate Shh activity in the control of molar tooth number. Development 2009, 136, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Tobin, J.L.; Beales, P.L. Bardet-Biedl syndrome: Beyond the cilium. Pediatr. Nephrol. 2007, 22, 926–936. [Google Scholar] [CrossRef]
- Ou, G.; Blacque, O.E.; Snow, J.J.; Leroux, M.R.; Scholey, J.M. Functional coordination of intraflagellar transport motors. Nature 2005, 436, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kantaputra, P.; Dejkhamron, P.; Sittiwangkul, R.; Katanyuwong, K.; Ngamphiw, C.; Sonsuwan, N.; Intachai, W.; Tongsima, S.; Beales, P.L.; Buranaphatthana, W. Dental Anomalies in Ciliopathies: Lessons from Patients with BBS2, BBS7, and EVC2 Mutations. Genes 2023, 14, 84. https://doi.org/10.3390/genes14010084
Kantaputra P, Dejkhamron P, Sittiwangkul R, Katanyuwong K, Ngamphiw C, Sonsuwan N, Intachai W, Tongsima S, Beales PL, Buranaphatthana W. Dental Anomalies in Ciliopathies: Lessons from Patients with BBS2, BBS7, and EVC2 Mutations. Genes. 2023; 14(1):84. https://doi.org/10.3390/genes14010084
Chicago/Turabian StyleKantaputra, Piranit, Prapai Dejkhamron, Rekwan Sittiwangkul, Kamornwan Katanyuwong, Chumpol Ngamphiw, Nuntigar Sonsuwan, Worrachet Intachai, Sissades Tongsima, Philip L. Beales, and Worakanya Buranaphatthana. 2023. "Dental Anomalies in Ciliopathies: Lessons from Patients with BBS2, BBS7, and EVC2 Mutations" Genes 14, no. 1: 84. https://doi.org/10.3390/genes14010084