Neuronopathic Gaucher disease: Beyond lysosomal dysfunction
 Nohela B. Arévalo1,2,3
Nohela B. Arévalo1,2,3  Cristian M. Lamaizon2,3 Viviana A. Cavieres4,5
Cristian M. Lamaizon2,3 Viviana A. Cavieres4,5  Patricia V. Burgos4,5,6
Patricia V. Burgos4,5,6  Alejandra R. Álvarez2,3,5 María J. Yañez7* Silvana Zanlungo1*
Alejandra R. Álvarez2,3,5 María J. Yañez7* Silvana Zanlungo1*- 1Department of Gastroenterology, Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Department of Cell and Molecular Biology, Biological Sciences Faculty, Pontificia Universidad Católica de Chile, Santiago, Chile
- 3Departamento de Biología Celular y Molecular, Facultad de Ciencias Biológicas, Millennium Institute on Immunology and Immunotherapy, Pontificia Universidad Católica, Santiago, Chile
- 4Facultad de Medicina y Ciencia, Centro de Biología Celular y Biomedicina (CEBICEM), Universidad San Sebastián, Santiago, Chile
- 5Facultad de Ciencias Biológicas, Centro de Envejecimiento y Regeneración (CARE-UC), Pontificia Universidad Católica, Santiago, Chile
- 6Centro Ciencia & Vida, Fundación Ciencia & Vida, Santiago, Chile
- 7Faculty of Medicine and Science, School of Medical Technology, Universidad San Sebastian, Concepción, Chile
Gaucher disease (GD) is an inherited disorder caused by recessive mutations in the GBA1 gene that encodes the lysosomal enzyme β-glucocerebrosidase (β-GC). β-GC hydrolyzes glucosylceramide (GluCer) into glucose and ceramide in the lysosome, and the loss of its activity leads to GluCer accumulation in different tissues. In severe cases, enzymatic deficiency triggers inflammation, organomegaly, bone disease, and neurodegeneration. Neuronopathic Gaucher disease (nGD) encompasses two different forms of the disease, characterized by chronic or acute damage to the central nervous system (CNS). The cellular and molecular studies that uncover the pathological mechanisms of nGD mainly focus on lysosomal dysfunction since the lysosome is the key organelle affected in GD. However, new studies show alterations in other organelles that contribute to nGD pathology. For instance, abnormal accumulation of GluCer in lysosomes due to the loss of β-GC activity leads to excessive calcium release from the endoplasmic reticulum (ER), activating the ER-associated degradation pathway and the unfolded protein response. Recent evidence indicates mitophagy is altered in nGD, resulting in the accumulation of dysfunctional mitochondria, a critical factor in disease progression. Additionally, nGD patients present alterations in mitochondrial morphology, membrane potential, ATP production, and increased reactive oxygen species (ROS) levels. Little is known about potential dysfunction in other organelles of the secretory pathway, such as the Golgi apparatus and exosomes. This review focuses on collecting evidence regarding organelle dysfunction beyond lysosomes in nGD. We briefly describe cellular and animal models and signaling pathways relevant to uncovering the pathological mechanisms and new therapeutic targets in GD.
Introduction
Gaucher disease (GD) is the most common lysosomal storage disorder (LSD), with a prevalence of 1.75:100,000 people worldwide (Nalysnyk et al., 2017; Institute of Gaucher Disease, 2020) and a higher incidence in populations with specific genetic backgrounds, such as Ashkenazi Jews (1:800) (Rosenbloom and Weinreb, 2013; Stirnemann et al., 2017).
GD pathology results mainly from recessive mutations in the GBA1 gene. This gene encodes the lysosomal enzyme β-glucocerebrosidase (β-GC) that hydrolyzes glucosylceramide (GluCer) into glucose and ceramide (Brady et al., 1966; Rosenbloom and Weinreb, 2013). There are more than 300 mutations that lead to a deficiency or insufficient activity of β-GC, which induces GluCer accumulation in the lysosomes (Hruska et al., 2008; Database The Human Genome, 2020). Macrophages are the most commonly affected cells in GD, called Gaucher cells (Lee, 1968). These cells are characterized by nuclear displacement, lysosomal swelling, and cytoplasm with a crumpled paper appearance (Lee, 1968). However, GluCer accumulation is also observed in macrophages from the reticuloendothelial system (such as Kupffer cells) (Brady, 1997; Simpson et al., 2014; Degnan et al., 2019) and neurons (Farfel-Becker et al., 2014).
There is also an abnormal accumulation of secondary lipids in addition to GluCer. For example, there is an increase in the deacetylated form of GluCer, Glucosylsphingosine (GluSph), in patient's cerebral and cerebellar cortices and GD animal models (Nilsson and Svennerholm, 1982; Orvisky et al., 2002; Dekker et al., 2011; Stiles et al., 2021). This buildup can start as early as conception and is linked to genetic severity (Orvisky et al., 2002). The GluSph toxic buildup is thought to cause neuronal death in GD2 via neurophagy (Nilsson and Svennerholm, 1982). For example, a study on LA-N-2 cells found that increasing GluSph treatment distorts neuronal morphology, declining neurite growth and acetylcholine production (Schueler et al., 2003). Similarly, it has been shown that GluSph can cause abnormal cell consequences such as inflammation, α-synuclein (α-syn) accumulation, and altered cell viability (Revel-Vilk et al., 2020). Although the exact mechanism by which GluSph contributes to GD2 disease is unknown, it is thought that GluSph blocks β-GC activity, establishing a negative feedback mechanism (Schueler et al., 2003; Revel-Vilk et al., 2020). In addition, cholesterol buildup has been linked to GD (Puri et al., 1999; Salvioli et al., 2005; Ron and Horowitz, 2008; Yañez et al., 2020). Fibroblasts from GD patients treated with inhibitors of cholesterol production reduce endoplasmic reticulum-associated responses and improve β-GC stability, maturation, and localization (Ron and Horowitz, 2008). Furthermore, cholesterol accumulation disrupts membrane traffic in the endocytic pathway in LSD, implying that secondary lipid accumulation contributes to GD pathogenesis. The abnormal accumulation of lipids in lysosomes results in clinical manifestations such as splenomegaly, hepatomegaly, bone disease, hematological alterations, and, in severe cases, neurodegeneration (Stirnemann et al., 2017). Indeed, the central nervous system (CNS) involvement has been a key determinant of GD classification. GD type 1 (GD1, OMIM #230800), the most prevalent form covering 90–95% of GD cases, is characterized by systemic manifestations (Stirnemann et al., 2017). GD type 2 (GD2, OMIM #230800) and GD type 3 (GD3, OMIM #230900) show CNS damage and are known as neuronopathic GD (hereafter nGD, unless its distinction is stated) (Stirnemann et al., 2017).
GD2 is the severe and progressive neuronopathic form of GD that manifest prenatally or during the first 9 months of life (Mignot et al., 2006). Although it contributes to <5% of cases, this form has a poor prognosis with non-effective treatment, and usually, children die before 2 years of age, with an average life expectancy of 11.7 months (Mignot et al., 2006). The low residual β-GC activity (10–15%) gives rise to the GD2 triad that encompasses neck rigidity, swallowing disorders, and oculomotor paralysis, along with systemic manifestations of the GD1 form (Weiss et al., 2015). Furthermore, GD2 could be accompanied by hydrops fetalis, in which fluids accumulate in different regions; however, these cases are perinatal lethal (Weiss et al., 2015). Furthermore, due to altered ceramide/glucosylceramide ratios in the skin, children present congenital ichthyosis. Organomegaly can be detected at birth (Weiss et al., 2015). This enlargement can be severe and affect feeding and breathing (Weiss et al., 2015).
Nevertheless, the prominent characteristic is the neurological decline due to a severe and rapidly progressive brainstem degeneration in the foreground (Mignot et al., 2006; Weiss et al., 2015). Patients frequently present the GD2 triad with arthrogryposis and microcephaly hypokinesia (Mignot et al., 2006; Weiss et al., 2015). The mechanism that leads to CNS dysfunction in GD2 is unknown; however, it is well-known that GluCer accumulation in neurons exceeds a threshold after which the inflammatory process and cell death begin (Farfel-Becker et al., 2014). This phenotype is characterized by microglial activation, astrocytosis, and neuronal death in layers III and V of the cortex and the CA2-CA4 hippocampal regions (Wong et al., 2004; Farfel-Becker et al., 2011, 2014). GD2 models present synaptic dysfunction, reduced synaptic size (Ginns et al., 2014), attenuated long-term potentiation (Sun et al., 2010), and alterations in synaptic proteins (Rocha et al., 2015).
GD3 (OMIM #231000) corresponds to a chronic neuronopathic form and encompasses 5–33% of GD cases (Stirnemann et al., 2017). The first manifestations start before 2 years of age (Tylki-Szymańska et al., 2010) but have a better prognosis than GD2 since patients can live over three decades (Schwartz et al., 2018). Oculomotor symptoms, cognitive deterioration, and seizures are the primary manifestation of CNS dysfunction; these symptoms occur along with systemic alterations.
Studying nGD remains a challenge to researchers since there is no clear relationship between the genotype and phenotype (Goker-Alpan et al., 2003). Although there are common mutations for specific subtypes, the same modifications could give rise to differences in severity, especially if there is CNS involvement (Goker-Alpan et al., 2003). Moreover, GBA1 mutations are one of the main risk factors for developing Parkinson's disease (PD) (Aflaki et al., 2017). It is still a matter of debate whether GD patients share pathophysiological characteristics, such as α-syn aggregation (Aflaki et al., 2017), with PD patients since some authors have shown that α-syn inclusion is present in GD patients with parkinsonism (Wong et al., 2004), while others did not detect α-syn inclusions in GD2 brains (Berger-Sieczkowski et al., 2016). In this regard, the relationship between GD and PD is an emerging and intriguing topic.
Cellular and molecular studies of nGD focus mainly on lysosomal dysfunction. However, several studies have reported that alterations in other cell organelles can also contribute to the pathology. For example, excessive calcium release from the endoplasmic reticulum (ER) leads to overactivation of ER-associated degradation (ERAD) and the unfolded protein response (UPR). Likewise, alterations in mitochondrial membrane potential, adenosine triphosphate (ATP) levels, and reactive oxygen species (ROS) have been observed. In this review, we focused on collecting and discussing the evidence regarding different dysfunctional organelles in GD. We review the knowledge considering the nGD model's main limitations and how this affects the interpretation of the results to uncover pathological mechanisms in nGD.
An overview of animal and cellular models of GD
The interpretation of results emerging from various cellular and animal models presenting a broad spectrum of pathological changes and differences is a major challenge in studying nGD. The first models of GD pathology started with the discovery of the selective and irreversible β-GC inhibitor conduritol-β-epoxide (CβE). Kanfer et al. used intraperitoneal and subcutaneous doses of CβE to treat mice. These animals present β-GC activity lower than 10%, showing the accumulation of GluCer in the brain, liver, and spleen (Kanfer et al., 1982). Variations in the treatment duration (Vardi et al., 2016), starting age of treatment (Marshall et al., 2002; Xu et al., 2008; Vardi et al., 2016), and genetic background were tested to improve GD characteristics (Klein et al., 2016; Vardi et al., 2016). Different genetic backgrounds treated with CβE presented distinct disease severity and progression (Klein et al., 2016). This model bias can complicate the interpretation of the results.
Neuronal lines and primary cultures treated with CβE have been used to generate GD cellular models. These pharmacological models are widely used since they are easy and fast to create and exhibit the main features of GD. Monocytes from GD patients have been used as a GD genetic model; however, their use is limited because of their low growth capacity (Aflaki et al., 2014; Bettman et al., 2015). Nevertheless, using the CRISPR/CAS9 technique, GBA1 silencing was triggered in the monocyte cell line THP-1 and glioblastoma U87 (Pavan et al., 2020). Both lines showed β-GC activation of <1% accompanied by accumulation of GluCer (Pavan et al., 2020). In addition, particular disease characteristics were reproduced, such as enzyme retention in the endoplasmic reticulum (ER) and activation of the response to misfolded proteins (UPR) (Pavan et al., 2020). Skin fibroblasts are also used as a genetic model of GD; however, these are not the primary cell type affected in nGD (Danes and Bearn, 1968). Nevertheless, it has been possible to obtain different cell types affected by the disease, such as osteoclasts (Panicker et al., 2018), macrophages (Panicker et al., 2012, 2014; Aflaki et al., 2014; Messelodi et al., 2021), and neurons (Schöndorf et al., 2014) from induced pluripotent stem cells (iPSCs) derived from GD fibroblasts (Santos and Tiscornia, 2017). Furthermore, the immortalization of cortical neurons from GBA1−/− mouse embryos provides a new tool for studying nGD (Westbroek et al., 2016). These GD neurons show a severe loss of more than 90% β-GC activity, GluCer accumulation, enlarged lysosomes, and altered calcium homeostasis compared with GBA1+/+ controls (Westbroek et al., 2016). These cell models are summarized in Table 1.

Table 1. Cellular models designed for GD study.
The first GBA1 knock-out mice generated showed <4% β-GC. This animal model showed GluCer accumulation mimicking nGD characteristics; nevertheless, these mice died 1 day after birth (Table 1) (Tybulewicz et al., 1992). Later, two mice with GBA1 point mutations were generated by inserting the recombinant mutation RecNcil or the L444P mutation (Liu et al., 1998) (Table 1). The L444P model was further improved by introducing a point mutation in the gene that encodes glucosylceramide synthase to attenuate GluCer synthesis (Mizukami et al., 2002). These mice displayed lower β-GC activity levels but did not present Gaucher cell characteristics (Table 1) (Mizukami et al., 2002). Xu et al. also generated mice models with GBA1 homozygous point mutations or heterozygous point mutations/null alleles (N370S, V394L, D409H, and D409V). Some of these animals survived longer than 60 weeks and presented Gaucher cells; however, none developed CNS involvement, confirming the defects in the brain are caused by GluCer accumulation (these and other models are summarized in Table 1) (Xu et al., 2003).
Several strategies have been used to eradicate β-GC activity in tissues, such as hematopoietic or mesenchymal cells (Enquist et al., 2006; Sinclair et al., 2007; Mistry et al., 2010). One such strategy was the development of GD2 conditional models. The first model presents a loss of β-GC activity in all tissues, except epidermal tissue (Enquist et al., 2007). The second model presents a loss of β-GC activity restricted to neuronal and glial progenitors (Enquist et al., 2007). These animal models present neuronopathic symptoms such as abnormal gait, hyperextension of the neck, and seizures with microglial activation and neuronal cell loss (Enquist et al., 2007). Unlike GBA1 KO mice, these conditional models have a longer life expectancy of 20–35 days, which provides an opportunity to study molecular pathways or treatments for more extended periods.
A new model of the chronic neuronopathic form was recently developed through the insertion of a Gba transgene regulated by a doxycycline system (Pewzner-Jung et al., 2021). The mice presented decreased β-GC activity levels in many brain areas and the liver, with GluCer accumulation and related motor and behavioral symptoms (Pewzner-Jung et al., 2021). This model resembles a GD3 phenotype with a longer survival period (10 months) and is proposed as an ideal model for studying the GD and PD relationship.
Since the link between GD and PD is an emerging topic, new GD models have been developed to uncover this relationship. A GBA1 non-sense mutant in medaka fish presents a 50% reduction in β-GC activity associated with neuronal pathology, showing progressive cell loss (TH+ neurons), microglial activation, and α-syn accumulation (Uemura et al., 2015). Additionally, mutations in the GBAb ortholog in D. melanogaster led to decreased β-GC activity, substrate accumulation, lysosomal swelling, activation of the unfolded protein response (UPR), and inflammatory pathways (Cabasso et al., 2019). These studies show the development of potential models for GD research that can resemble neuropathology and improve life span (Table 2).
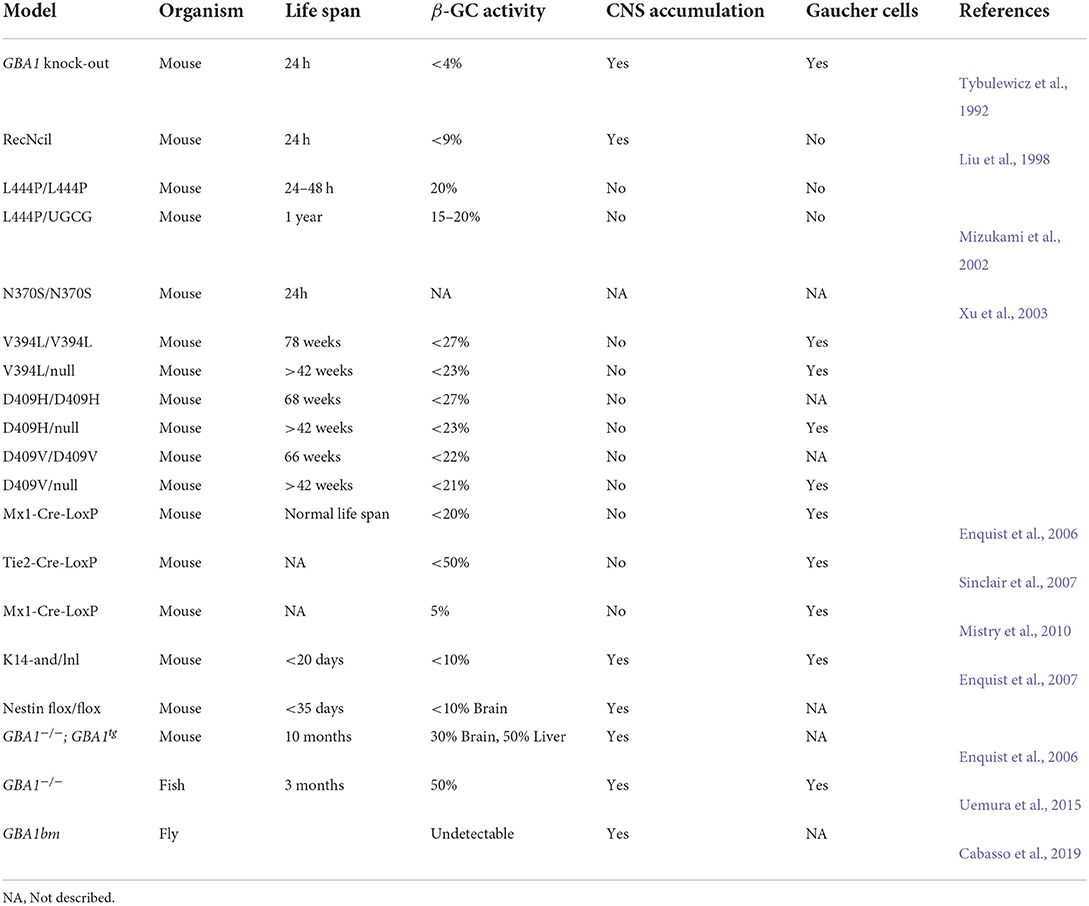
Table 2. Animal models designed for GD study.
Signaling pathways related to organelle dysfunction in nGD
The loss of β-GC activity and accumulation of GluCer trigger lysosomal dysfunction, impacting the function of other organelles due in part to alterations in specific signaling pathways. For instance, iPSC/GD-neuronal progenitors and IPCS/GD-neurons from GD2 patients show hyperactivity of the mammalian target of rapamycin complex 1 (mTORC1) kinase, which inhibits the function of the lysosomal master transcription factor TFEB (Brown et al., 2019). This transcription factor is responsible for inducing the expression of several lysosomal biogenesis and autophagy-related genes (Brown et al., 2019). The treatment of IPCS/GD-neurons with torin 1, a mTORC1 inhibitor, induces TFEB nuclear translocation, thus promoting lysosomal biogenesis, increasing LC3II/LAMP1 association, and decreasing p62 levels (Brown et al., 2019; Srikanth et al., 2021).
Furthermore, the necroptotic cell death pathway could be activated in nGD (Vitner et al., 2014; Yañez et al., 2021). In a genetic mouse model of GD2, neuronal cell death was not associated with increased apoptosis markers, but rather with elevated mRNA and protein levels of RIPK1 and RIPK3 – two kinases involved in necroptotic cell death (Vitner et al., 2014). Interestingly, RIPK3 knock-out mice exhibited decreased inflammatory signs in the CNS and improved motor symptoms and mouse lifespan (Vitner et al., 2014). Likewise, there was RIPK3 activation in different pharmacological and genetic nGD models, including patient fibroblasts (Yañez et al., 2021), CβE-treated neuronal lines, and nGD mice, which supported the key role of RIPK3 in nGD pathology (Yañez et al., 2021).
Interestingly, c-Abl tyrosine kinase is activated in different GD models. c-Abl signaling has been involved in the pathogenic mechanisms of several lysosomal storage disorders (LSDs). For instance, c-Abl is activated in Niemann–Pick C (NPC), a disease characterized by cholesterol accumulation in lysosomes. In NPC models, c-Abl signaling leads to increased p73 pro-apoptotic protein levels (Klein et al., 2011), amyloid precursor protein (APP) amyloidogenic processing (Yáñez et al., 2016), increases in histone deacetylase 2 (HDAC2) levels, repression of neuronal and synaptic genes, and retention of TFEB in the cytoplasm (Gonzalez-Zuñiga et al., 2014). Moreover, the c-Abl pharmacological inhibition in NPC models reduced cerebellar apoptosis and promoted autophagy and cholesterol clearance—changes associated with increased TFEB nuclear localization, TFEB target gene expression, lysosomal biogenesis, and exocytosis (Contreras et al., 2020). Recently, we found that the inhibition of c-Abl is also involved in alterations in the autophagy lysosomal pathway in Niemann–Pick A (NPA), a disease in which sphingomyelin accumulates in lysosomes (Marín et al., 2022). The inhibition of c-Abl improves autophagy lysosomal function, restoring autophagy flux and sphingomyelin accumulation in NPA models (Marín et al., 2022). These results show that alterations in c-Abl signaling are relevant in LSDs and suggest that it could also play an essential pathogenic role in GD.
Interestingly, in GD models, there is c-Abl activation and crosstalk between c-Abl and the RIPK1/RIPK3/MLKL pathway (Yañez et al., 2021). c-Abl interacts with RIPKP3, and its genetic inhibition decreases RIPKP3 levels. c-Abl mediates RIPK3 tyrosine phosphorylation in both GD neuronal model and fibroblasts from GD patients. These results suggest that c-Abl could play an important role in GD pathology by regulating RIPK3 signaling.
Thus, lysosome dysfunction and lysosome-altered signaling pathways are central to GD pathology. Nevertheless, the lysosome is a dynamic organelle. Its functions impact the function of other organelles through direct and indirect signaling pathways and membrane contact sites (MCSs) that dynamically regulate the morphology, fusion–fission events, distribution, and organelle function. Therefore, the disruption of lysosome integrity, such as β-GC deficiency and GluCer accumulation, will affect other organelles beyond just the lysosome.
Dysregulation of organelles in nGD
Lysosomal dysfunction: The central axis of nGD pathology
The lysosome is the most affected organelle in GD since β-GC deficiency and GluCer accumulation alter the lysosome number, distribution, pH, enzymatic content, and activity (Figure 1). For instance, neuroblastoma cells with a GBA1 mutation show an abnormal increase in acidic punctate structures positive to lysotracker staining; this could be associated with a higher number of acidic compartments (Schöndorf et al., 2014; García-Sanz et al., 2017). Likewise, iPSC-derived neurons from GBA1-associated PD patients showed an increased number and size of LAMP1-positive structures (Schöndorf et al., 2014; García-Sanz et al., 2017). One possible interpretation is that cells may respond to increasing lysosomal biogenesis to compensate for the lysosomal dysfunction induced by the loss of β-GC activity. Alternatively, the increased number of LAMP1-positive structures could indicate an accumulation of damaged lysosomes.
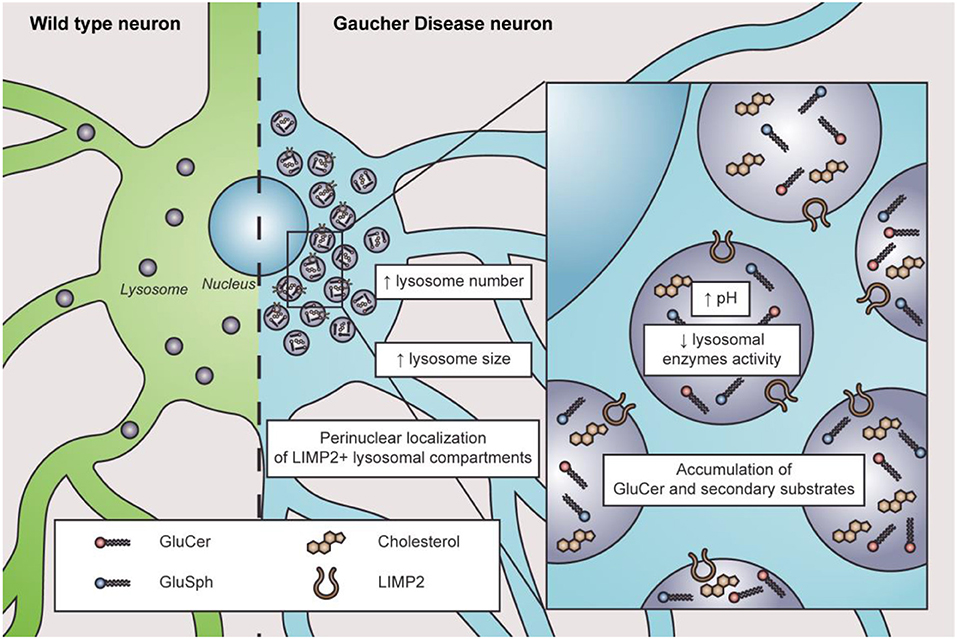
Figure 1. Characteristics of lysosome dysfunction in nGD. In physiological conditions, lysosomes are distributed throughout the cytosol in wild-type neurons (left in green). In nGD, lysosomes are increased in number and size with a perinuclear localization of LIMP2+ lysosomal compartments accompanied by elevated intraluminal pH and decreased lysosomal enzyme activity, along with the accumulation of GluCer and secondary substrates such as glucosylsphingosine (GluSph) and cholesterol (right in light blue).
Altered lysosome distribution with preferentially perinuclear distribution is an early characteristic of nGD (Awad et al., 2015, 2017; Zigdon et al., 2017). The cause of altered lysosome distribution is unclear. However, cytoskeletal impairments are related to altered lysosome distribution since microtubule stabilization with paclitaxel decreases the percentage of altered lysosome distribution in neurons treated with CβE (Zigdon et al., 2017). Moreover, in a GD2 model, only LIMP2-positive lysosomes are found in the perinuclear region, observing no changes regarding LAMP1-positive lysosomes (Zigdon et al., 2017), which suggests that different lysosome subtypes might exist in nGD. Interestingly, LIMP2 is responsible for β-GC trafficking to the lysosomes (Reczek et al., 2007), and its downregulation is associated with reduced β-GC activity (Thomas et al., 2021). Additionally, lysosome localization depends on their maturation state since immature lysosomes are preferentially distributed in the cytoplasm periphery, while mature lysosomes are more abundant in the perinuclear region (Pu et al., 2016; Oyarzún et al., 2019). Lysosomal pH plays a pivotal role in lysosomal enzymatic activities. In this sense, cathepsins and acid sphingomyelinase present lower enzymatic activity in many cellular models of nGD (Tatti et al., 2012; García-Sanz et al., 2017; Panicker et al., 2018; Polinski et al., 2021). Indeed, a higher lysosomal pH from 5.5 to 6 was reported in astrocytes with GBA1 mutations, RAW macrophages treated with CβE, and lymphoblasts derived from GD patients (Sillence, 2013). The increased pH in GD pathology is remarkable since the change in lysosomal pH in other LSDs is subtle (Sillence, 2013). Accordingly, defects in lysosome exocytosis were observed in osteoclasts and iPSCs from nGD patients, which was also related to lower membrane repair capacity (Panicker et al., 2018).
The parallel alterations of the endoplasmic reticulum in nGD
The endoplasmic reticulum is the first station where β-GC transits and folds. Therefore, it is intuitive to think that the mutations could cause changes in its tertiary structure and folding, impacting the ER state. Endoplasmic reticulum stress, ERAD overactivation, and UPR response were reported in several nGD models (Figure 2).
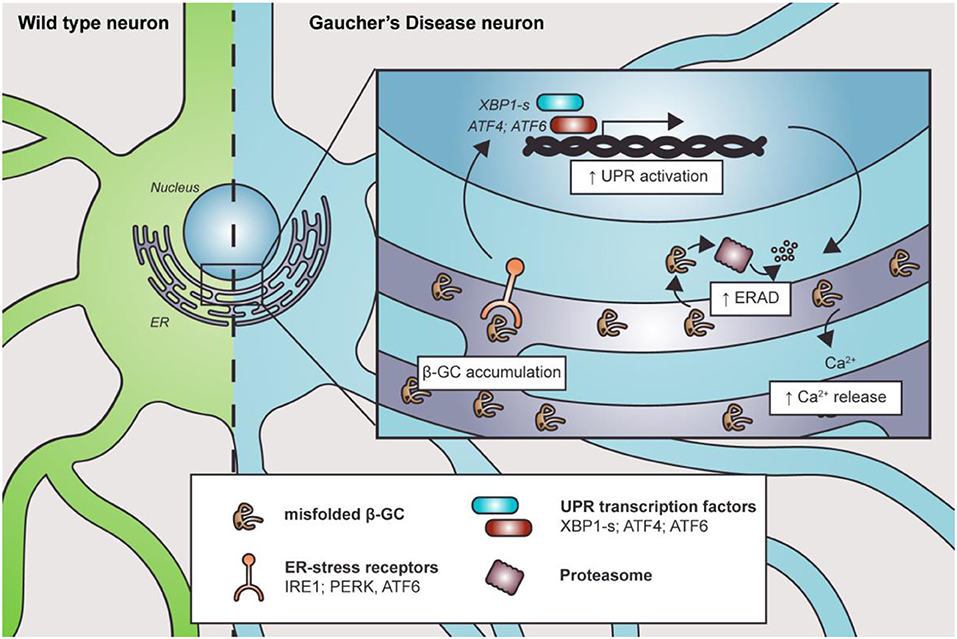
Figure 2. Misfolded β-GC mediates ER stress. Mutations in GBA1 lead to β-GC misfolding, promoting the activation of ER stress receptors and the UPR. This, in turn, induces degradation of the misfolded enzyme through ERAD. Along with this, Ca2+ homeostasis is also altered, contributing to cell and organelle damage.
The expression of human mutant β-GC was linked with ER defects and activation of UPR signaling in D. melanogaster (Maor et al., 2013a,b). Furthermore, the mutant forms of β-GC colocalize with ER–protein calnexin, a chaperone that participates in the pro-folding pathway and degradation of misfolded proteins, in fibroblasts from nGD patients (Maor et al., 2013a,b). Interestingly, the level of β-GC/calnexin colocalization is related to disease severity (Maor et al., 2013a,b). Similarly, the ubiquitin E3 ligase ITCH, known as atrophin-1-interacting protein 4, binds to mutant, but not to the wild-type, β-GC to mediate its ERAD degradation, another parameter that has been related to disease severity (Maor et al., 2013a,b).
In the D. melanogaster nGD model, the expression of human mutant β-GC leads to neurodevelopmental eye defects and UPR signaling activation (Suzuki et al., 2013). Exacerbated or prolonged misfolded protein accumulation could trigger ER stress and activation of the UPR (Suzuki et al., 2013). Accordingly, treatment with ambroxol, a pharmacological chaperone that promotes β-GC folding and trafficking, reverts ER stress and neurodevelopmental defects (Suzuki et al., 2013). Also, GD patients' fibroblasts and iPSCs carrying a heterozygous GBA1 mutation showed increased spliced xbp1 mRNA levels, upregulation of ER dBiP and CHOP proteins, and increased eIF2α phosphorylation (Wei et al., 2008; Maor et al., 2013b; Fernandes et al., 2016). The increase in dBip and xbp1 mRNA levels suggests the activation of the IRE1a branch of the UPR, probably due to ER stress. Additionally, the elevated levels of Xbp1 spliced mRNA could explain the upregulation of CHOP, a target of the XBP1s transcription factor. This branch could activate apoptosis in response to stress. The IRE1a/XBP1 branch of the UPR correlates with the expression of other proteins such as ERAD and ERAD-enhancing α-mannosidase-like proteins (EDEMs). Moreover, GD1 and GD2 fibroblasts showed increased Grp78/Bip, Grp94, ATF6, and SOD2 protein levels, which are important ER chaperones. Also, these cells are more susceptible to brefeldin-mediated apoptosis (Wei et al., 2008). The increased misfolded protein response is probably due to the accumulation of β-GC, which releases Grp78/Bip from the UPR receptor and activates different pathways, including the ATF6 UPR branch (Wei et al., 2008). One of the ATF6 target genes is Xbp1, which amplifies the mis-/unfolded protein response. Another ATF6 target gene is CHOP, suggesting that GD fibroblasts are more prone to cell death under stressful conditions (Wei et al., 2008).
Intriguingly, it seems that β-GC accumulation in the ER, and not lysosomal substrate accumulation, is the cause of UPR activation since β-GC inhibition with CβE does not lead to UPR activation in healthy fibroblasts (Maor et al., 2013b). UPR activation was also corroborated in D. melanogaster GD models (Maor et al., 2013b; Sanchez-Martinez et al., 2016). In this regard, the treatment of flies with an isofagomine, a chaperon that promotes protein folding, decreased Xbp-1 protein levels and improved the decline in climbing caused by the expression of mutant β-GC (Sanchez-Martinez et al., 2016). Moreover, Mu et al. showed promising results using the celastrol chaperone in skin GD fibroblasts carrying the L444P mutation, which increases β-GC activity, folding, and trafficking to the lysosome and avoids activation of the ERAD response. These effects are partially mediated by UPR activation since inhibition of ATF6, IRE1, and PERK blocks the effects of celastrol (Mu et al., 2008). These results open the possibility that at least some mutations found in the GBA1 gene could be treated by chemical chaperones or by ERAD enhancers to diminish the levels of misfolded β-GC.
ER stress could also be mediated by dysregulation of calcium (Ca2+) ER homeostasis. The first evidence of the relationship between β-GC deficit and Ca2+ alterations was described by Korkotian et al. (1999). These authors showed that β-GC inhibition with CβE in hippocampal neurons leads to a three-fold increase in ER volume, observing tubular membrane structures, accompanied by increased ryanodine receptor (RyR) levels and Ca2+ release after glutamate or caffeine treatment (Korkotian et al., 1999). Furthermore, CβE-treated cells were more sensitive to glutamate-induced neuronal cell death, which was blocked by pre-incubating with ryanodine. This suggests that increased Ca2+ release from the ER could contribute to neuronal death in neuronopathic forms of GD (Korkotian et al., 1999).
Pelled et al. showed that microsomes from the brains of GD2 patients had higher Ca2+ release than microsomes from GD1 patients, GD3 patients, and healthy subjects. Additionally, Ca2+ positively correlated with the level of GluCer accumulation in the same microsomes (Pelled et al., 2005). Furthermore, treatment with thapsigargin, an inhibitor of the ER-Ca2+ ATPase, induces an elevated release of Ca2+ in fibroblasts from GD1 patients and PD patients carrying GBA1 mutations (Kilpatrick et al., 2016). These facts support the idea that the accumulation of the misfolded enzyme in the ER could contribute to toxic GluCer accumulation in the lysosome (Kilpatrick et al., 2016). In conclusion, the ER is affected by nGD and contributes to cellular alterations. This opens the question of whether lysosomes are the central organelle affected by Gaucher disease or whether there is a parallel dysfunction in the ER due to the misfolded enzyme.
Autophagosome: Early and late failures of autophagy in nGD
Macroautophagy (hereafter referred to as autophagy) is necessary for proper cell function. There are two main types of autophagy: bulk autophagy, where the cytoplasmic material is recycled, and selective autophagy, where specific organelles or materials, such as aggregated proteins, are sequestered by the autophagy machinery to be degraded after its fusion with the lysosome (Ravikumar et al., 2009). Several studies have demonstrated increased autophagy markers, such as the lipidated forms of LC3, LC3-II, and SQSTM1/p62 in nGD models (Tatti et al., 2012; Schöndorf et al., 2014; Awad et al., 2015; Bae et al., 2015; Du et al., 2015; Aflaki et al., 2016; García-Sanz et al., 2017; Brown et al., 2019; Li et al., 2019). This increase in autophagy protein levels seems to be related to a defect in autophagy flux, rather than a consequence of increased autophagosome biogenesis. For instance, it has been proposed that GluCer accumulation leads to a direct decrease in autophagosome/lysosome fusion (Awad et al., 2015; Du et al., 2015) and, consequently, defects in autophagosome clearance and protein accumulation (Awad et al., 2015, 2017; Du et al., 2015). These defects in lysosomal degradation capacity and autophagic flux have also been observed in pharmacological and genetic nGD animal models. For example, mice treated with CβE present increased levels of SQSTM1/p62 in the substantia nigra and cortex, two main areas affected in GD (Rocha et al., 2015). Likewise, increased levels of these proteins were observed in mice with a GBA1 L444P heterozygous mutation, supporting the idea that deficient β-GC activity leads to autophagic defects (Li et al., 2019). Mice with mutations on GBA1 and Sap C—which encode the lysosomal β-GC activator saposin C—also present increased SQSTM1/p62 levels in the brain stem, basal ganglia, and thalamus. Similar results were observed in new models of D. melanogaster, which contain altered Ref(2)P and ATG8—homologs of p62/SQSTM1 and LC3, respectively (Davis et al., 2016).
In addition to substrate accumulation, protein degradation through autophagy and chaperone-mediated autophagy (CMA) is impaired in nGD (Sun and Grabowski, 2010). In line with this, α-syn is a protein degraded by both types of autophagy (Kuo et al., 2022). Some authors have not observed accumulation of its toxic status in the brains of infants with GD2, although altered plasma α-syn levels were reported (Berger-Sieczkowski et al., 2016). Additionally, other authors observed inclusions of α-syn in the brains of GD patients with parkinsonism (Wong et al., 2004), raising the question of whether α-syn accumulation is directly linked to β-GC activity in GD or is it a result of the parkinsonism present in some GD patients.
Several studies show that a reduction in β-GC stabilizes α-syn, promoting its aggregation (Manning-Bog et al., 2009; Mazzulli et al., 2011; Taguchi et al., 2017). However, the mechanisms by which β-GC deficiency promotes α-syn aggregation are controversial. Some authors suggest that GluCer accumulation itself could induce the formation of soluble oligomeric intermediates, thus influencing the amyloid formation of purified α-syn (Mazzulli et al., 2011). At the same time, others suggest that insufficient or mutated β-GC could promote the formation of these aggregates. Supporting the latter idea, neurons treated with CβE neither increase insoluble monomeric α-syn levels nor its phosphorylation status (Gegg and Schapira, 2016). Correctly folded β-GC can interact with α-syn in a way that promotes its degradation (Yap et al., 2015), while the GD-related mutation N370S could weaken this interaction, increasing the probability of α-syn aggregation (Yap et al., 2013). This toxic aggregation contributes to autophagy dysfunction, resulting in a bidirectional loop since toxic α-syn can block β-GC trafficking to lysosomes. By contrast, the autophagy machinery, including the ATG8-independent CMA, cannot degrade these toxic aggregates (Mazzulli et al., 2011; Kuo et al., 2022).
Along with lysosomal-related dysfunction in autophagy, initial autophagy steps were also altered. Accordingly, the hippocampus of L444P heterozygous mice showed elevated levels of LC3-II and SQSTM1/p62 compared with wild-type (WT) animals (de la Mata et al., 2015). Moreover, the treatment of GBA1 mutant neurons with the autophagy inducer rapamycin and autophagy flux blockers, leupeptin, and pepstatin diminishes the increase in autophagic vacuoles compared to WT neurons, indicating that both early and late autophagies are altered in GD (Li et al., 2019).
In another nGD model, LC3-II levels were reduced in GBA1−/− cells compared to WT cells. This observation could be due to impaired LC3 conjugation with phosphatidylethanolamine since ATG5/12 levels were also reduced. Consistent with this, astrocyte knock-out in GBA1 induces a diffuse cytoplasmic distribution of LC3 and impairs the formation of autophagic structures (Osellame et al., 2013). These differences in autophagy marker levels and LC3 distribution seem to be related to the extent of β-GC deficiency since GBA1+/− astrocytes can form autophagosomes (Osellame et al., 2013). Accordingly, no accumulation of β-GC substrates was detected in GBA+/− mouse models or the brains of PD patients with heterozygous GBA1 mutations (Sardi et al., 2011; Farfel-Becker et al., 2014; Gegg and Schapira, 2016).
Mitochondrial damage as a prominent feature in nGD
Altered mitochondrial homeostasis and its consequences are widely described in other neurodegenerative diseases and LSDs (Osellame and Duchen, 2014; Stepien et al., 2020). In nGD, some studies show altered mitochondrial morphology, membrane potential, and energetic metabolism. Specifically, decreased mitochondrial membrane potential (ΔΨm), fragmented mitochondria, reduced ATP production levels, and increased ROS production have been observed in different nGD models (Cleeter et al., 2013; Osellame et al., 2013; Dasgupta et al., 2015; de la Mata et al., 2015; Yun et al., 2018; Ivanova et al., 2019; Li et al., 2019; Morén et al., 2019). These effects were previously noticed in later treatment days with CβE in SH-SY5Y cells (Cleeter et al., 2013). Furthermore, GBA1−/− neurons and astrocytes showed increased mitochondrial mass due to blocked autophagy flux. Interestingly, the incubation of cells with MITOQ10, a mitochondrial antioxidant, does not change LC3 II protein levels, which are decreased in this model. This suggests that mitochondrial alterations are secondary effects of organelle dysfunction and defective degradation of these mitochondria via autophagy (Osellame et al., 2013). Additionally, dysfunctional mitochondria in GBA1−/− neurons and astrocytes could not recruit Parkin, a ubiquitin E3 ligase essential for marking mitochondria for autophagy degradation, and these cells were unable to form autophagic vesicles (Osellame et al., 2013).
By contrast, GD fibroblasts harboring L444P/L444P mutation presented elevated LC3 II, a block in the autophagic flux, and accumulated autophagic vesicles containing depolarized mitochondria. This evidence suggests that mitophagy activation and impaired autophagic flux are present in this GD model (de la Mata et al., 2015). Furthermore, these differences in altered mitophagy regulation seem to be related to the specific Gaucher model since GBA1+/− astrocytes, but not GBA1−/−, can form autophagic vacuoles that could sequester dysfunctional mitochondria (Osellame et al., 2013).
Thus far, it is unknown whether mitochondrial dysfunction is a direct consequence of lysosomal dysfunction or if other cellular events contribute. Recently, Kim et al. showed that β-GC mutations in dopaminergic iPSC-derived neurons harboring heterozygous GBA1 mutations led to prolonged mitochondria–lysosome (M-L) contact sites compared to isogenic controls. This correlated with decreased mitochondrial density in axons. Increased proteasomal degradation of TBC1C15—a Rab7 GAP, which facilitates GTP hydrolysis and mitochondria–lysosome untethering—could cause these prolonged contacts (Kim et al., 2021). Interestingly, this altered mitochondria–lysosome dynamic was reproduced in the pharmacologic nGD model, and restoring β-GC function retrieved this long mitochondria–lysosome contact phenotype. Additionally, inhibition of other lysosomal enzymes does not disrupt mitochondria–lysosome contacts in neurons, suggesting that β-GC and GluCer exert a pivotal role in mitochondria–lysosome dynamics (Kim et al., 2021).
A greater colocalization coefficient of β-amyloid precursor protein (APP) and α-syn inclusions with the mitochondrial proteins TOM40 and COX IV were observed in cortical neurons of an nGD mouse model (Xu et al., 2014). By contrast, a small portion of these proteins colocalized with autophagosome markers LC3 II and p62 or with the lysosome (LAMP2) (Xu et al., 2014). These results suggest preferential clustering of APP/α-syn in mitochondrial compartments in cortical GD neurons, showing electron-dense and enlarged mitochondria with functional defects (Xu et al., 2014). The altered influx of APP/α-syn could block mitochondrial protein import channels, leading to the arrest of essential proteins for mitochondrial function (Devi et al., 2006). The chronic neuroinflammatory environment and decreased antioxidant compounds in GD could play a pivotal role in mitochondrial dysfunction.
Emerging evidence of organelle dysfunction in GD pathology
Although the lysosome, ER, and mitochondria are the prominent organelles explored in nGD pathology, other organelles could also play a pivotal role. In fibroblasts harboring the N370S GBA1 mutation, the Golgi apparatus (GA) alterations are striking, showing a fragmented structure, dispersed into small elements. Compared to control cells, heterozygous GBA1 mutants have shorter and smaller GA cisternae (García-Sanz et al., 2017). Alteration of the Golgi apparatus structure could have functional consequences. For instance, inhibition of β-GC by CβE alters lactosylceramide sorting from the GA to the lysosome, suggesting that loss of β-GC activity impacts GA-related traffic, which could be related to alterations in its structure or functional components (Sillence et al., 2002).
Likewise, exosomes have gained increased attention since they participate in cellular communication and signaling in a bidirectional way in health and disease. Interestingly, plasma from GD patients presents large and multilayered exosomes with various morphologies compared to healthy controls (Tatiana et al., 2020). Some of these exosomes were electron-dense and had a higher fluorescence intensity of exosome markers CD9 and CD81 than controls (Tatiana et al., 2020). In this regard, Papadopoulos et al. showed that β-GC inhibition increases the number of brain exosomes and α-syn-associated exosomes in an overexpressing α-syn mouse model treated with CβE. Although further studies are required to establish the exosome role in GD pathology, they may have a relevant role as a garbage transporter for clearing or transporting signal molecules that can contribute to or decrease the pathological environment (Papadopoulos et al., 2018).
Conclusion
This review aimed to evaluate the functional consequences of lysosomal dysfunction on other organelles, such as the ER and mitochondria (Figure 3).
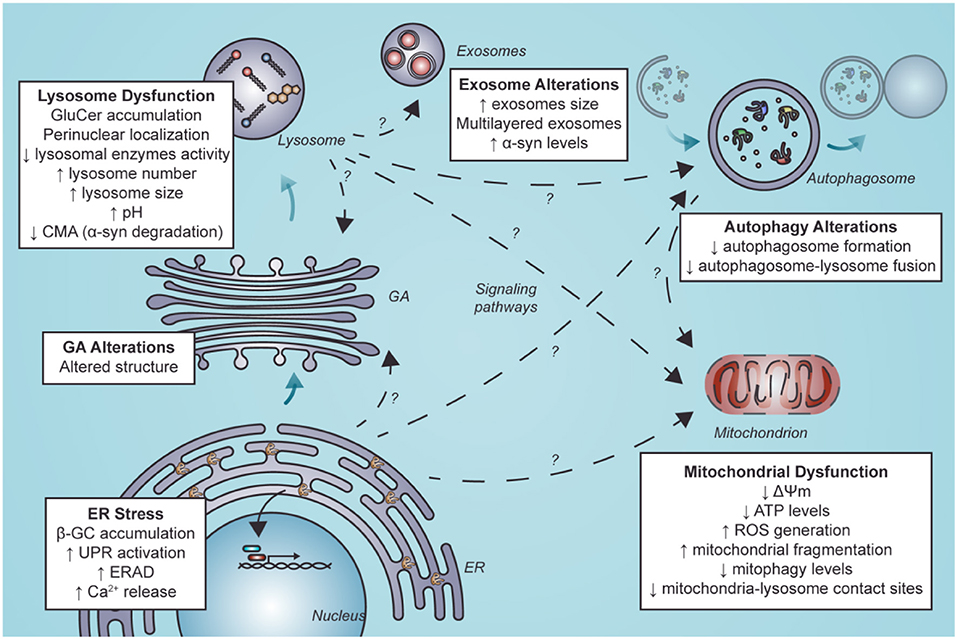
Figure 3. Secondary organelle dysfunction in nGD following ER stress and lysosome dysfunction. Lysosome dysfunction is characterized by GluCer accumulation, perinuclear localization of LIMP2-positive structures, diminished lysosomal enzymes activity, elevated lysosome number and size, alkalinization of lysosomes and decreased levels of CMA. Furthermore, GluCer accumulation and ER stress mediated by β-GC misfolding can alter other organelles. These alterations could be mediated by direct interaction with other organelles. They could also result from indirect interactions between lysosomes and other organelles by activating signaling pathways or alterations in these organelles such as exosomes, autophagosomes in autophagy, mitochondria, and GA. Alterations in autophagy and mitochondria have been well described. Still, little is known about the mechanisms that lead to these alterations and less about alterations in other organelles such as exosomes and GA (segmented arrows with a question mark). GluCer, glucosylceramide; CMA, chaperone-mediated autophagy; ER, endoplasmic reticulum; β-GC, β-glucocerebrosidase; GA, Golgi apparatus; ERAD, endoplasmic reticulum-associated degradation; ROS, reactive oxygen species.
nGD is an inherited recessive autosomal metabolic defect due to deficiency of the lysosomal enzyme β-GC that induces neurodegeneration. Decreased catalytic activity and instability of β-GC lead to the accumulation of glucosylceramide, glucosylsphingosine, and cholesterol in the lysosomes of macrophage cells and visceral organs, predominantly in the liver, spleen, and bone marrow.
Dysfunction of the autophagy–lysosomal pathway represents a key pathogenic event in GBA1-associated neurodegeneration. The autophagy–lysosomal pathway maintains cellular homeostasis by clearing protein aggregates and damaged organelles, a process critical for neuronal survival. However, the pathological consequences are not only due to the intrinsic β-GC enzymatic dysfunction but also the consequence of abnormalities occurring during its transport and delivery to the lysosome. Thus, β-GC misfolding during its passage through the ER can lead to premature degradation by the proteasome, activating ER stress receptors and the UPR. Along with this, Ca2+ homeostasis is also altered, contributing to cell and ER damage. Furthermore, mutant forms of β-GC colocalize with calnexin (Ron and Horowitz, 2005), and there is a significant correlation between disease severity and the level of β-CG calnexin interaction (Ron and Horowitz, 2005). In line with this, the reduction of the β-CG degradation rate promotes its delivery to the pro-folding calnexin pathway and increases enzymatic activity in Gaucher disease fibroblasts (Tan et al., 2014).
On the other hand, mitochondrial dysfunction is emerging as a significant contributor to the pathophysiology of lysosomal storage disorders. Lysosomes are essential for autophagy and autophagic clearance of dysfunctional mitochondria and represent an essential element of mitochondrial quality control. We suggest that the compromised mitochondrial function in these cells may increase the risk of neurodegeneration in neurons that are already vulnerable because of their normal physiological activity.
Although lysosomal dysfunction is key to the loss of cell homeostasis in nGD, other organelles could be affected as a direct or indirect consequence of lysosome perturbation. In this regard, modulation of specific mitochondrial and endoplasmic reticulum pathways could be a new therapeutic frontier in GD management. We propose that other cell components such as GA and exosomes could play a role yet to be uncovered in nGD pathology.
Author contributions
NA, AÁ, MY, and SZ: writing—original draft preparation. NA, CL, VC, PB, AÁ, MY, and SZ: conceptualization and writing—review and editing. AÁ, MY, and SZ: funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Agencia Nacional de Investigación y Desarrollo (ANID), FONDECYT [Grants Nos. Q221201668 (AÁ), 11200592 (MY), and 1190334 (SZ)], FONDEF ID21I10347, Millennium Science Initiative Program ICN 09_016/ICN 2021_045, Millennium Institute on Immunology and Immunotherapy, and Centro de Envejecimiento y Regeneración CARE-Chile-UC Center under grant number AFB170005 (AÁ and PB).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aflaki, E., Moaven, N., Borger, D. K., Lopez, G., Westbroek, W., Chae, J. J., et al. (2016). Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 15, 77–88. doi: 10.1111/acel.12409
Aflaki, E., Stubblefield, B. K., Maniwang, E., Lopez, G., Moaven, N., Goldin, E., et al. (2014). Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci. Transl. Med. 6, 240ra73. doi: 10.1126/scitranslmed.3008659
Aflaki, E., Westbroek, W., and Sidransky, E. (2017). The complicated relationship between gaucher disease and parkinsonism: insights from a rare disease. Neuron 93, 737–746. doi: 10.1016/j.neuron.2017.01.018
Awad, O., Panicker, L. M., Deranieh, R. M., Srikanth, M. P., Brown, R. A., Voit, A., et al. (2017). Altered differentiation potential of Gaucher's Disease iPSC neuronal progenitors due to Wnt/β-catenin downregulation. Stem Cell Rep. 9, 1853–1867. doi: 10.1016/j.stemcr.2017.10.029
Awad, O., Sarkar, C., Panicker, L. M., Miller, D., Zeng, X., Sgambato, J. A., et al. (2015). Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 24, 5775–5788. doi: 10.1093/hmg/ddv297
Bae, E. J., Yang, N. Y., Lee, C., Lee, H. J., Kim, S., Sardi, S. P., et al. (2015). Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 47, e153. doi: 10.1038/emm.2014.128
Berger-Sieczkowski, E., Lutz, M. I., Auff, E., and Kovacs, G. G. (2016). Gaucher cells are not associated with α-synuclein neuropathology in infants. Clin. Neuropathol. 35, 122. doi: 10.5414/NP300901
Bettman, N., Avivi, I., Rosenbaum, H., Bisharat, L., and Katz, T. (2015). Impaired migration capacity in monocytes derived from patients with Gaucher disease. Blood Cells Mol. Dis. 55, 180–186. doi: 10.1016/j.bcmd.2014.12.003
Brady, R. O. (1997). 1 Gaucher's disease: past, present and future. Baillières Clin. Haematol. 10, 621–634. doi: 10.1016/S0950-3536(97)80031-5
Brady, R. O., Kanfer, J. N., Bradley, R. M., and Shapiro, D. (1966). Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher's disease. J. Clin. Invest. 45, 1112–1115. doi: 10.1172/JCI105417
Brown, R. A., Voit, A., Srikanth, M. P., Thayer, J. A., Kingsbury, T. J., Jacobson, M. A., et al. (2019). mTOR hyperactivity mediates lysosomal dysfunction in Gaucher's disease iPSC-neuronal cells. Dis. Models Mech. 12, dmm038596. doi: 10.1242/dmm.038596
Cabasso, O., Paul, S., Dorot, O., Maor, G., Krivoruk, O., Pasmanik-Chor, M., et al. (2019). Drosophila melanogaster mutated in its GBA1b ortholog recapitulates neuronopathic gaucher disease. J. Clin. Med. 8, 1420. doi: 10.3390/jcm8091420
Cleeter, M. W., Chau, K. Y., Gluck, C., Mehta, A., Hughes, D. A., Duchen, M., et al. (2013). Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 62, 1–7. doi: 10.1016/j.neuint.2012.10.010
Contreras, P. S., Tapia, P. J., González-Hódar, L., Peluso, I., Soldati, C., Napolitano, G., et al. (2020). c-Abl inhibition activates TFEB and promotes cellular clearance in a lysosomal disorder. iScience 23, 101691. doi: 10.1016/j.isci.2020.101691
Danes, B. S., and Bearn, A. G. (1968). Gaucher's disease: a genetic disease detected in skin fibroblast cultures. Science 161, 1347–1348. doi: 10.1126/science.161.3848.1347
Dasgupta, N., Xu, Y. H., Li, R., Peng, Y., Pandey, M. K., Tinch, S. L., et al. (2015). Neuronopathic Gaucher disease: dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 24, 7031–7048. doi: 10.1093/hmg/ddv404
Database The Human Genome (2020). GBA Mutations Database. Database The Human Genome. Available online at: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GBA.
Davis, M. Y., Trinh, K., Thomas, R. E., Yu, S., Germanos, A. A., Whitley, B. N., et al. (2016). Glucocerebrosidase deficiency in drosophila results in α-synuclein-independent protein aggregation and neurodegeneration. PLoS Genet. 12, e1005944. doi: 10.1371/journal.pgen.1005944
de la Mata, M., Cotán, D., Oropesa-Ávila, M., Garrido-Maraver, J., Cordero, M. D., Villanueva Paz, M., et al. (2015). Pharmacological chaperones and coenzyme Q10 Treatment improves mutant β-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of Gaucher disease. Sci. Rep. 5, 10903. doi: 10.1038/srep10903
Degnan, A. J., Ho-Fung, V. M., Ahrens-Nicklas, R. C., Barrera, C. A., Serai, S. D., Wang, D. J., et al. (2019). Imaging of non-neuronopathic Gaucher disease: recent advances in quantitative imaging and comprehensive assessment of disease involvement. Insights Imaging 10, 70. doi: 10.1186/s13244-019-0743-5
Dekker, N., van Dussen, L., Hollak, C. E., Overkleeft, H., Scheij, S., Ghauharali, K., et al. (2011). Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood 118, e118–e127. doi: 10.1182/blood-2011-05-352971
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Du, T. T., Wang, L., Duan, C. L., Lu, L. L., Zhang, J. L., Gao, G., et al. (2015). GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 11, 1803–1820. doi: 10.1080/15548627.2015.1086055
Enquist, I. B., Lo Bianco, C., Ooka, A., Nilsson, E., Månsson, J. E., Ehinger, M., et al. (2007). Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 104, 17483–17488. doi: 10.1073/pnas.0708086104
Enquist, I. B., Nilsson, E., Ooka, A., Månsson, J. E., Olsson, K., Ehinger, M., et al. (2006). Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 103, 13819–13824. doi: 10.1073/pnas.0606016103
Farfel-Becker, T., Vitner, E. B., Kelly, S. L., Bame, J. R., Duan, J., Shinder, V., et al. (2014). Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 23, 843–854. doi: 10.1093/hmg/ddt468
Farfel-Becker, T., Vitner, E. B., Pressey, S. N. R., Eilam, R., Cooper, J. D., Futerman, A. H., et al. (2011). Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum. Mol. Genet. 20, 1375–1386. doi: 10.1093/hmg/ddr019
Fernandes, H. J. R., Hartfield, E. M., Christian, H. C., Emmanoulidou, E., Zheng, Y., Booth, H., et al. (2016). ER Stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson's iPSC-derived dopamine neurons. Stem Cell Rep. 6, 342–356. doi: 10.1016/j.stemcr.2016.01.013
García-Sanz, P., Orgaz, L., Bueno-Gil, G., Espadas, I., Rodríguez-Traver, E., Kulisevsky, J., et al. (2017). N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson's disease. Move. Disord. 32, 1409–1422. doi: 10.1002/mds.27119
Gegg, M. E., and Schapira, A. H. (2016). Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol. Dis. 90, 43–50. doi: 10.1016/j.nbd.2015.09.006
Ginns, E. I., Mak, S. K. K., Ko, N., Karlgren, J., Akbarian, S., Chou, V. P., et al. (2014). Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 111, 152–162. doi: 10.1016/j.ymgme.2013.12.003
Goker-Alpan, O., Schiffmann, R., Park, J. K., Stubblefield, B. K., Tayebi, N., Sidransky, E., et al. (2003). Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J. Pediatr. 143, 273–276. doi: 10.1067/S0022-3476(03)00302-0
Gonzalez-Zuñiga, M., Contreras, P. S., Estrada, L. D., Chamorro, D., Villagra, A., Zanlungo, S., et al. (2014). c-Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer's disease. Mol. Cell. 56, 163–173. doi: 10.1016/j.molcel.2014.08.013
Hruska, K. S., LaMarca, M. E., Scott, C. R., and Sidransky, E. (2008). Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 29, 567–583. doi: 10.1002/humu.20676
Institute of Gaucher Disease (2020). Epidemiology of Gaucher Disease. Institute of Gaucher Disease. Available online at: https://www.gaucher-institute.com/burden-of-disease/epidemiology-of-gaucher-disease
Ivanova, M. M., Changsila, E., Iaonou, C., and Goker-Alpan, O. (2019). Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 14, e0210617. doi: 10.1371/journal.pone.0210617
Kanfer, J. N., Stephens, M. C., Singh, H., and Legler, G. (1982). The Gaucher mouse. Prog. Clin. Biol. Res. 95, 627–644.
Kilpatrick, B. S., Magalhaes, J., Beavan, M. S., McNeill, A., Gegg, M. E., Cleeter, M. W. J., et al. (2016). Endoplasmic reticulum and lysosomal Ca2? stores are remodelled in GBA1-linked Parkinson disease patient fibroblasts. Cell Calcium. 59, 12–20. doi: 10.1016/j.ceca.2015.11.002
Kim, S., Wong, Y. C., Gao, F., and Krainc, D. (2021). Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson's disease. Nat. Commun. 12, 1807. doi: 10.1038/s41467-021-22113-3
Klein, A., Maldonado, C., Vargas, L. M., Gonzalez, M., Robledo, F., de Perez, K. P., et al. (2011). Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiol. Dis. 41, 209–218. doi: 10.1016/j.nbd.2010.09.008
Klein, A. D., Ferreira, N. S., Ben-Dor, S., Duan, J., Hardy, J., Cox, T. M., et al. (2016). Identification of modifier genes in a mouse model of Gaucher disease. Cell Rep. 16, 2546–2553. doi: 10.1016/j.celrep.2016.07.085
Korkotian, E., Schwarz, A., Pelled, D., Schwarzmann, G., Segal, M., Futerman, A. H., et al. (1999). Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem. 274, 21673–21678. doi: 10.1074/jbc.274.31.21673
Kuo, S. H., Tasset, I., Cheng Melody, M., Diaz, A., Pan, M. K., Lieberman Ori, J., et al. (2022). Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 8, eabm6393. doi: 10.1126/sciadv.abm6393
Lee, R. E. (1968). The fine structure of the cerebroside occurring in Gaucher's disease. Proc. Natl. Acad. Sci. U.S.A. 61, 484–489. doi: 10.1073/pnas.61.2.484
Li, H., Ham, A., Ma, T. C., Kuo, S. H., Kanter, E., Kim, D., et al. (2019). Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 15, 113–130. doi: 10.1080/15548627.2018.1509818
Liu, Y., Suzuki, K., Reed, J. D., Grinberg, A., Westphal, H., Hoffmann, A., et al. (1998). Mice with type 2 and 3 Gaucher disease point mutations generated by a single insertion mutagenesis procedure. Proc. Natl. Acad. Sci. U.S.A. 95, 2503–2508. doi: 10.1073/pnas.95.5.2503
Manning-Bog, A. B., Schüle, B., and Langston, J. W. (2009). Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 30, 1127–1132. doi: 10.1016/j.neuro.2009.06.009
Maor, G., Filocamo, M., and Horowitz, M. (2013a). ITCH regulates degradation of mutant glucocerebrosidase: implications to Gaucher disease. Hum. Mol. Genet. 22, 1316–1327. doi: 10.1093/hmg/dds535
Maor, G., Rencus-Lazar, S., Filocamo, M., Steller, H., Segal, D., Horowitz, M., et al. (2013b). Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet J. Rare Dis. 8, 140. doi: 10.1186/1750-1172-8-140
Marín, T., Dulcey, A. E., Campos, F., de la Fuente, C., Acuña, M., Castro, J., et al. (2022). c-Abl activation linked to autophagy-lysosomal dysfunction contributes to neurological impairment in niemann-pick type A disease. Front. Cell Dev. Biol. 10, 844297. doi: 10.3389/fcell.2022.844297
Marshall, J., McEachern, K. A., Kyros, J. A., Nietupski, J. B., Budzinski, T., Ziegler, R. J., et al. (2002). Demonstration of feasibility of in vivo gene therapy for Gaucher disease using a chemically induced mouse model. Mol. Ther. 6, 179–189. doi: 10.1006/mthe.2002.0650
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Messelodi, D., Bertuccio, S. N., Indio, V., Strocchi, S., Taddia, A., Serravalle, S., et al. (2021). iPSC-Derived gaucher macrophages display growth impairment and activation of inflammation-related cell death. Cells 10, 2822. doi: 10.3390/cells10112822
Mignot, C., Doummar, D., Maire, I., De Villemeur, T. B., and French Type 2 Gaucher Disease Study Group. (2006). Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 28, 39–48. doi: 10.1016/j.braindev.2005.04.005
Mistry, P. K., Liu, J., Yang, M., Nottoli, T., McGrath, J., Jain, D., et al. (2010). Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. U.S.A. 107, 19473–19478. doi: 10.1073/pnas.1003308107
Mizukami, H., Mi, Y., Wada, R., Kono, M., Yamashita, T., Liu, Y., et al. (2002). Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Invest. 109, 1215–1221. doi: 10.1172/JCI0214530
Morén, C., Juárez-Flores, D. L., Chau, K. Y., Gegg, M., Garrabou, G., González-Casacuberta, I., et al. (2019). GBA mutation promotes early mitochondrial dysfunction in 3D neurosphere models. Aging 11, 10338–10355. doi: 10.18632/aging.102460
Mu, T. W., Ong, D. S., Wang, Y. J., Balch, W. E., Yates, J. R., Segatori, L., et al. (2008). Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 134, 769–781. doi: 10.1016/j.cell.2008.06.037
Nalysnyk, L., Rotella, P., Simeone, J. C., Hamed, A., and Weinreb, N. (2017). Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology 22, 65–73. doi: 10.1080/10245332.2016.1240391
Nilsson, O., and Svennerholm, L. (1982). Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 39, 709–718. doi: 10.1111/j.1471-4159.1982.tb07950.x
Orvisky, E., Park, J. K., LaMarca, M. E., Ginns, E. I., Martin, B. M., Tayebi, N., et al. (2002). Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol. Genet. Metab. 76, 262–270. doi: 10.1016/S1096-7192(02)00117-8
Osellame, L. D., and Duchen, M. R. (2014). Quality control gone wrong: mitochondria, lysosomal storage disorders and neurodegeneration. Br. J. Pharmacol. 171, 1958–1972. doi: 10.1111/bph.12453
Osellame, L. D., Rahim, A. A., Hargreaves, I. P., Gegg, M. E., Richard-Londt, A., Brandner, S., et al. (2013). Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson's disease. Cell Metab. 17, 941–953. doi: 10.1016/j.cmet.2013.04.014
Oyarzún, J. E., Lagos, J., Vázquez, M. C., Valls, C., De la Fuente, C., Yuseff, M. I., et al. (2019). Lysosome motility and distribution: relevance in health and disease. Biochim. Biophys. Acta. Mol. Basis Dis. 1865, 1076–1087. doi: 10.1016/j.bbadis.2019.03.009
Panicker, L. M., Miller, D., Awad, O., Bose, V., Lun, Y., Park, T. S., et al. (2014). Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells 32, 2338–2349. doi: 10.1002/stem.1732
Panicker, L. M., Miller, D., Park, T. S., Patel, B., Azevedo, J. L., Awad, O., et al. (2012). Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 109, 18054–18059. doi: 10.1073/pnas.1207889109
Panicker, L. M., Srikanth, M. P., Castro-Gomes, T., Miller, D., Andrews, N. W., Feldman, R. A., et al. (2018). Gaucher disease iPSC-derived osteoblasts have developmental and lysosomal defects that impair bone matrix deposition. Hum. Mol. Genet. 27, 811–822. doi: 10.1093/hmg/ddx442
Papadopoulos, V. E., Nikolopoulou, G., Antoniadou, I., Karachaliou, A., Arianoglou, G., Emmanouilidou, E., et al. (2018). Modulation of β-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson's disease. Hum. Mol. Genet. 27, 1696–1710. doi: 10.1093/hmg/ddy075
Pavan, E., Ormazabal, M., Peruzzo, P., Vaena, E., Rozenfeld, P., Dardis, A., et al. (2020). CRISPR/Cas9 editing for Gaucher disease modelling. Int. J. Mol. Sci. 21, 3268. doi: 10.3390/ijms21093268
Pelled, D., Trajkovic-Bodennec, S., Lloyd-Evans, E., Sidransky, E., Schiffmann, R., Futerman, A. H., et al. (2005). Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol. Dis. 18, 83–88. doi: 10.1016/j.nbd.2004.09.004
Peng, Y., Liou, B., Lin, Y., Fannin, V., Zhang, W., Feldman, R. A., et al. (2021). Substrate reduction therapy reverses mitochondrial, mtor, and autophagy alterations in a cell model of gaucher disease. Cells 10, 2286. doi: 10.3390/cells10092286
Pewzner-Jung, Y., Joseph, T., Blumenreich, S., Vardi, A., Ferreira, N. S., Cho, S. M., et al. (2021). Brain pathology and cerebellar purkinje cell loss in a mouse model of chronic neuronopathic Gaucher disease. Prog. Neurobiol. 197, 101939. doi: 10.1016/j.pneurobio.2020.101939
Polinski, N. K., Martinez, T. N., Gorodinsky, A., Gareus, R., Sasner, M., Herberth, M., et al. (2021). Decreased glucocerebrosidase activity and substrate accumulation of glycosphingolipids in a novel GBA1 D409V knock-in mouse model. PLoS ONE 16, e0252325. doi: 10.1371/journal.pone.0252325
Pu, J., Guardia, C. M., Keren-Kaplan, T., and Bonifacino, J. S. (2016). Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339. doi: 10.1242/jcs.196287
Puri, V., Watanabe, R., Dominguez, M., Sun, X., Wheatley, C. L., Marks, D. L., et al. (1999). Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol. 1, 386–388. doi: 10.1038/14084
Ravikumar, B., Futter, M., Jahreiss, L., Korolchuk, V. I., Lichtenberg, M., Luo, S., et al. (2009). Mammalian macroautophagy at a glance. J. Cell. Sci. 122 (Pt. 11), 1707–1711. doi: 10.1242/jcs.031773
Reczek, D., Schwake, M., Schröder, J., Hughes, H., Blanz, J., Jin, X., et al. (2007). LIMP-2 Is a receptor for lysosomal mannose-6-phosphate-independent targeting of β-glucocerebrosidase. Cell 131, 770–783. doi: 10.1016/j.cell.2007.10.018
Revel-Vilk, S., Fuller, M., and Zimran, A. (2020). Value of glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher disease: a systematic literature review. Int. J. Mol. Sci. 21, 7159. doi: 10.3390/ijms21197159
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Graham, A. R., Brown, E., et al. (2015). Sustained systemic glucocerebrosidase inhibition induces brain α-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid. Redox Signal. 23, 550–564. doi: 10.1089/ars.2015.6307
Ron, I., and Horowitz, M. (2005). ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 14, 2387–2398. doi: 10.1093/hmg/ddi240
Ron, I., and Horowitz, M. (2008). Intracellular cholesterol modifies the ERAD of glucocerebrosidase in Gaucher disease patients. Mol. Genet. Metab. 93, 426–436. doi: 10.1016/j.ymgme.2007.10.132
Rosenbloom, B. E., and Weinreb, N. J. (2013). Gaucher disease: a comprehensive review. Crit. Rev. Oncog. 18, 163–175. doi: 10.1615/CritRevOncog.2013006060
Salvioli, R., Tatti, M., Scarpa, S., Moavero, S. M., Ciaffoni, F., Felicetti, F., et al. (2005). The N370S (Asn370–>Ser) mutation affects the capacity of glucosylceramidase to interact with anionic phospholipid-containing membranes and saposin C. Biochem. J. 390 (Pt. 1), 95–103. doi: 10.1042/BJ20050325
Sanchez-Martinez, A., Beavan, M., Gegg, M. E., Chau, K. Y., Whitworth, A. J., Schapira, A. H., et al. (2016). Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 6, 31380. doi: 10.1038/srep31380
Santos, D. M., and Tiscornia, G. (2017). Induced pluripotent stem cell modeling of gaucher's disease: what have we learned? Int. J. Mol. Sci. 18, 888. doi: 10.3390/ijms18040888
Sardi, S. P., Clarke, J., Kinnecom, C., Tamsett, T. J., Li, L., Stanek, L. M., et al. (2011). CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. U.S.A. 108, 12101–12106. doi: 10.1073/pnas.1108197108
Schöndorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5, 4028. doi: 10.1038/ncomms5028
Schueler, U. H., Kolter, T., Kaneski, C. R., Blusztajn, J. K., Herkenham, M., Sandhoff, K., et al. (2003). Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: a model system for assessing neuronal damage in Gaucher disease type 2 and 3. Neurobiol. Dis. 14, 595–601. doi: 10.1016/j.nbd.2003.08.016
Schwartz, I. V. D., Göker-Alpan, Ö., Kishnani, P. S., Zimran, A., Renault, L., Panahloo, Z., et al. (2018). Characteristics of 26 patients with type 3 Gaucher disease: a descriptive analysis from the Gaucher outcome survey. Mol. Genet. Metab. Rep. 14, 73–79. doi: 10.1016/j.ymgmr.2017.10.011
Sillence, D. J. (2013). Glucosylceramide modulates endolysosomal pH in Gaucher disease. Mol. Genet. Metab. 109, 194–200. doi: 10.1016/j.ymgme.2013.03.015
Sillence, D. J., Puri, V., Marks, D. L., Butters, T. D., Dwek, R. A., Pagano, R. E., et al. (2002). Glucosylceramide modulates membrane traffic along the endocytic pathway. J. Lipid Res. 43, 1837–1845. doi: 10.1194/jlr.M200232-JLR200
Simpson, W. L., Hermann, G., and Balwani, M. (2014). Imaging of Gaucher disease. World J. Radiol. 6, 657–668. doi: 10.4329/wjr.v6.i9.657
Sinclair, G. B., Jevon, G., Colobong, K. E., Randall, D. R., Choy, F. Y., Clarke, L. A., et al. (2007). Generation of a conditional knockout of murine glucocerebrosidase: utility for the study of Gaucher disease. Mol. Genet. Metab. 90, 148–156. doi: 10.1016/j.ymgme.2006.09.008
Srikanth, M. P., Jones, J. W., Kane, M., Awad, O., Park, T. S., Zambidis, E. T., et al. (2021). Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1. Stem Cells Transl. Med. 10, 1081–1094. doi: 10.1002/sctm.20-0386
Stepien, K. M., Roncaroli, F., Turton, N., Hendriksz, C. J., Roberts, M., Heaton, R. A., et al. (2020). Mechanisms of mitochondrial dysfunction in lysosomal storage disorders: a review. J. Clin. Med. 9, 2596. doi: 10.3390/jcm9082596
Stiles, A. R., Huggins, E., Fierro, L., Jung, S. H., Balwani, M., Kishnani, P. S., et al. (2021). The role of glucosylsphingosine as an early indicator of disease progression in early symptomatic type 1 Gaucher disease. Mol. Genet. Metab. Rep. 27, 100729. doi: 10.1016/j.ymgmr.2021.100729
Stirnemann, J., Belmatoug, N., Camou, F., Serratrice, C., Froissart, R., Caillaud, C., et al. (2017). A review of gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 18, 441. doi: 10.3390/ijms18020441
Sun, Y., and Grabowski, G. A. (2010). Impaired autophagosomes and lysosomes in neuronopathic Gaucher disease. Autophagy 6, 648–649. doi: 10.4161/auto.6.5.12047
Sun, Y., Liou, B., Ran, H., Skelton, M. R., Williams, M. T., Vorhees, C. V., et al. (2010). Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 19, 1088–1097. doi: 10.1093/hmg/ddp580
Suzuki, T., Shimoda, M., Ito, K., Hanai, S., Aizawa, H., Kato, T., et al. (2013). Expression of human Gaucher disease gene GBA generates neurodevelopmental defects and ER stress in Drosophila eye. PLoS ONE 8, e69147. doi: 10.1371/journal.pone.0069147
Taguchi, Y. V., Liu, J., Ruan, J., Pacheco, J., Zhang, X., Abbasi, J., et al. (2017). Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated Parkinson's disease. J. Neurosci. 37, 9617–9631. doi: 10.1523/JNEUROSCI.1525-17.2017
Tan, Y. L., Genereux, J. C., Pankow, S., Aerts, J. M. F. G., Yates, J. R. III., and Kelly, J. W. (2014). ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher's disease. Chem. Biol. 21, 967–976. doi: 10.1016/j.chembiol.2014.06.008
Tatiana, S., Stanislav, N., Darya, K., Luiza, G., Konstantin, S., Sergey, L., et al. (2020). Altered level of plasma exosomes in patients with Gaucher disease. Eur. J. Med. Genet. 63, 104038. doi: 10.1016/j.ejmg.2020.104038
Tatti, M., Motta, M., Di Bartolomeo, S., Scarpa, S., Cianfanelli, V., Cecconi, F., et al. (2012). Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum. Mol. Genet. 21, 5159–5173. doi: 10.1093/hmg/dds367
Thomas, R., Moloney, E. B., Macbain, Z. K., Hallett, P. J., and Isacson, O. (2021). Fibroblasts from idiopathic Parkinson's disease exhibit deficiency of lysosomal glucocerebrosidase activity associated with reduced levels of the trafficking receptor LIMP2. Mol. Brain 14, 16. doi: 10.1186/s13041-020-00712-3
Tybulewicz, V. L. J., Tremblay, M. L., LaMarca, E. L., Willemsen, R., Stubblefield, B. K., Winfield, S., et al. (1992). Animal model of Gaucher's disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 357, 407–410. doi: 10.1038/357407a0
Tylki-Szymańska, A., Vellodi, A., El-Beshlawy, A., Cole, J. A., and Kolodny, E. (2010). Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the international collaborative gaucher group neurological outcomes subregistry. J. Inherit. Metab. Dis. 33, 339–346. doi: 10.1007/s10545-009-9009-6
Uemura, N., Koike, M., Ansai, S., Kinoshita, M., Ishikawa-Fujiwara, T., Matsui, H., et al. (2015). Viable neuronopathic Gaucher disease model in Medaka (Oryzias latipes) displays axonal accumulation of alpha-synuclein. PLoS Genet. 11, e1005065. doi: 10.1371/journal.pgen.1005065
Vardi, A., Zigdon, H., Meshcheriakova, A., Klein, A. D., Yaacobi, C., Eilam, R., et al. (2016). Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J. Pathol. 239, 496–509. doi: 10.1002/path.4751
Vitner, E. B., Salomon, R., Farfel-Becker, T., Meshcheriakova, A., Ali, M., Klein, A. D., et al. (2014). RIPK3 as a potential therapeutic target for Gaucher's disease. Nat. Med. 20, 204–208. doi: 10.1038/nm.3449
Wei, H., Kim, S. J., Zhang, Z., Tsai, P. C., Wisniewski, K. E., Mukherjee, A. B., et al. (2008). ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum. Mol. Genet. 17, 469–477. doi: 10.1093/hmg/ddm324
Weiss, K., Gonzalez, A., Lopez, G., Pedoeim, L., Groden, C., Sidransky, E., et al. (2015). The clinical management of Type 2 Gaucher disease. Mol. Genet. Metab. 114, 110–122. doi: 10.1016/j.ymgme.2014.11.008
Westbroek, W., Nguyen, M., Siebert, M., Lindstrom, T., Burnett, R. A., Aflaki, E., et al. (2016). A new glucocerebrosidase-deficient neuronal cell model provides a tool to probe pathophysiology and therapeutics for Gaucher disease. Dis. Model. Mech. 9, 769–778. doi: 10.1242/dmm.024588
Wong, K., Sidransky, E., Verma, A., Mixon, T., Sandberg, G. D., Wakefield, L. K., et al. (2004). Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 82, 192–207. doi: 10.1016/j.ymgme.2004.04.011
Xu, Y. H., Quinn, B., Witte, D., and Grabowski, G. A. (2003). Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am. J. Pathol. 163, 2093–2101. doi: 10.1016/S0002-9440(10)63566-3
Xu, Y. H., Reboulet, R., Quinn, B., Huelsken, J., Witte, D., Grabowski, G. A., et al. (2008). Dependence of reversibility and progression of mouse neuronopathic Gaucher disease on acid β-glucosidase residual activity levels. Mol. Genet. Metab. 94, 190–203. doi: 10.1016/j.ymgme.2008.01.013
Xu, Y. H., Xu, K., Sun, Y., Liou, B., Quinn, B., Li, R. H., et al. (2014). Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 23, 3943–3957. doi: 10.1093/hmg/ddu105
Yáñez, M. J., Belbin, O., Estrada, L. D., Leal, N., Contreras, P. S., Lleó, A., et al. (2016). c-Abl links APP-BACE1 interaction promoting APP amyloidogenic processing in Niemann-pick type C disease. Biochim. Biophys. Acta. 1862, 2158–2167. doi: 10.1016/j.bbadis.2016.08.016
Yañez, M. J., Campos, F., Marín, T., Klein, A. D., Futerman, A. H., Alvarez, A. R., et al. (2021). c-Abl activates RIPK3 signaling in Gaucher disease. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166089. doi: 10.1016/j.bbadis.2021.166089
Yañez, M. J., Marín, T., Balboa, E., Klein, A. D., Alvarez, A. R., Zanlungo, S., et al. (2020). Finding pathogenic commonalities between niemann-pick type C and other lysosomal storage disorders: opportunities for shared therapeutic interventions. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165875. doi: 10.1016/j.bbadis.2020.165875
Yap, T. L., Jiang, Z., Heinrich, F., Gruschus, J. M., Pfefferkorn, C. M., Barros, M., et al. (2015). Structural features of membrane-bound glucocerebrosidase and α-synuclein probed by neutron reflectometry and fluorescence spectroscopy. J. Biol. Chem. 290, 744–754. doi: 10.1074/jbc.M114.610584
Yap, T. L., Velayati, A., Sidransky, E., and Lee, J. C. (2013). Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol. Genet. Metab. 108, 56–64. doi: 10.1016/j.ymgme.2012.11.010
Yun, S. P., Kim, D., Kim, S., Kim, S., Karuppagounder, S. S., Kwon, S. H., et al. (2018). α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 13, 1. doi: 10.1186/s13024-017-0233-5
Keywords: lysosomal storage disorders (LSD), Neuronopathic Gaucher disease (nGD), Parkinson's disease (PD), lysosome, autophagy, endoplasmic reticulum, mitochondria, Golgi apparatus
Citation: Arévalo NB, Lamaizon CM, Cavieres VA, Burgos PV, Álvarez AR, Yañez MJ and Zanlungo S (2022) Neuronopathic Gaucher disease: Beyond lysosomal dysfunction. Front. Mol. Neurosci. 15:934820. doi: 10.3389/fnmol.2022.934820
Received: 03 May 2022; Accepted: 27 June 2022;
Published: 03 August 2022.
Edited by:
Stefanie Flunkert, QPS Austria, AustriaReviewed by:
Dan-Lucian Dumitraşcu, Iuliu Haţieganu University of Medicine and Pharmacy, RomaniaAdeline Lau, Flinders University, Australia
Emanuele Frattini, University of Milan, Italy
Copyright © 2022 Arévalo, Lamaizon, Cavieres, Burgos, Álvarez, Yañez and Zanlungo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María J. Yañez, maria.yanez@uss.cl; Silvana Zanlungo, szanlungo@uc.cl