Impaired Mitochondrial Mobility in Charcot-Marie-Tooth Disease
 Cara R. Schiavon1,2
Cara R. Schiavon1,2  Uri Manor
Uri Manor- 1Waitt Advanced Biophotonics Center, Salk Institute for Biological Studies, La Jolla, CA, United States
- 2Molecular and Cell Biology Laboratory, Salk Institute for Biological Studies, La Jolla, CA, United States
Charcot-Marie-Tooth (CMT) disease is a progressive, peripheral neuropathy and the most commonly inherited neurological disorder. Clinical manifestations of CMT mutations are typically limited to peripheral neurons, the longest cells in the body. Currently, mutations in at least 80 different genes are associated with CMT and new mutations are regularly being discovered. A large portion of the proteins mutated in axonal CMT have documented roles in mitochondrial mobility, suggesting that organelle trafficking defects may be a common underlying disease mechanism. This review will focus on the potential role of altered mitochondrial mobility in the pathogenesis of axonal CMT, highlighting the conceptional challenges and potential experimental and therapeutic opportunities presented by this “impaired mobility” model of the disease.
Introduction
Charcot-Marie-Tooth (CMT) disease is the most commonly inherited neurological disorder, affecting ∼1 in 5000 people (Skre, 1974; Barreto et al., 2016). It is a peripheral neuropathy defined by progressive deterioration of the peripheral nerves in the distal parts of the body, specifically the feet, hands, and lower extremities. This typically results in both motor and sensory loss in the affected areas. Unraveling the pathogenic mechanism(s) underlying CMT is somewhat complicated by the fact that CMT is both genetically and clinically heterogeneous. CMT has many subtypes including, demyelinating (affecting mainly Schwann cells), axonal, and intermediate (affecting both axons and Schwann cells). Herein, we are focusing on perturbations of mitochondrial mobility that might underlie the pathogenesis of axonal CMT.
Charcot-Marie-Tooth variants were originally classified based purely on clinical data. However, a recent explosion in genetic data can be mined to generate some compelling hypotheses. To date, over 100 mutations across more than 40 different proteins have been implicated in axonal and intermediate CMT. A large fraction of CMT-associated proteins has been shown or is predicted to affect the mobility of mitochondria or other organelles (Table 1). In this review, we will focus on those that affect mitochondrial mobility and hypothesize that defects in this process might begin to explain why CMT mutations mainly affect peripheral neurons. At the same time, we highlight important limitations to this “impaired mobility” model. Our belief is that the insights gained from studying the effects of CMT mutations in peripheral neurons will inform the role of mitochondrial mobility in other types of neurons and neurodegenerative disorders, including those associated with aging.

Table 1. Genes mutated in axonal and intermediate CMT.
CMT Is a Progressive Disorder That Affects Predominantly the Longest Neurons
Charcot-Marie-Tooth usually affects only the feet, hands, and lower extremities. The axons leading to these distal sites can be as long as a meter in some individuals. There have been some reports of central nervous system involvement but these instances are rare (Pareyson and Marchesi, 2009; Lee et al., 2017). Some CMT mutations also cause optic atrophy, and the optical nerve notably consists of relatively long axons (∼50 mm). Patients are usually born unaffected, but typically by age 10 display major losses of function. However, the range of age can be from the toddler years to the 5th decade of life (Skre, 1974; Verhoeven et al., 2003; Zuchner et al., 2004, 2006; Chung et al., 2006; Engelfried et al., 2006; Cho et al., 2007; Calvo et al., 2009; Braathen et al., 2010; Boyer et al., 2011b). The severity of the disease is directly correlated with the age of onset (Chung et al., 2006; Verhoeven et al., 2006), and the longer axons (i.e., the feet) invariably degenerate before the shorter axons (i.e., the hands). In cases where CMT patients also display optic atrophy, this occurs after loss of function in the hands (Verhoeven et al., 2006; Zuchner et al., 2006). Together, these observations indicate a disease that directly correlates the length of the axon with the speed of onset, and the speed of onset with the magnitude of the pathology.
CMT Mutations Largely Affect Mitochondrial Mobility
As mentioned above, a unique peripheral nerve characteristic is their extreme length, which suggests these cells are uniquely sensitive to impaired long-distance transport. Put simply, a decrease in mobility would have a greater impact on longer distance commutes than shorter ones. In support of this theory, 23 out of the 48 genes mutated in axonal or intermediate CMT encode proteins that play roles in mitochondrial function, often impacting mitochondrial mobility (Table 1).
The majority of axonal CMT studies have centered on Mitofusin 2 (MFN2) mutations, which consistently result in reduced axonal mitochondrial mobility. This phenotype has been reproduced in mouse models and in patient cell lines and tissues. Neurons expressing MFN2 CMT mutants and neurons from MFN2 CMT mouse models also show reduced axonal mitochondrial mobility (Baloh et al., 2007; Vallat et al., 2008; Rocha et al., 2018). MFN2 is also implicated in mitochondrial fusion dynamics, and MFN2 CMT mutations cause clustering of improperly fused mitochondria (Baloh et al., 2007; Detmer and Chan, 2007; Vallat et al., 2008; Rocha et al., 2018). Thus, it is possible that this mitochondrial clustering contributes to reduced mitochondrial mobility. Although several MFN2 CMT mutants cause mitochondrial fragmentation suggesting a disruption of its fusogenic activity, there are other MFN2 CMT mutants that do not alter mitochondrial morphology or, seemingly paradoxically, even cause mitochondrial elongation (Detmer and Chan, 2007; Codron et al., 2016; Rocha et al., 2018). MFN2 is also implicated in mitophagy, lipid transfer, lipid droplet-mitochondria contacts, and endoplasmic reticulum (ER)-mitochondria contacts, although whether MFN2 increases or decreases ER-mitochondria contacts is still under debate (de Brito and Scorrano, 2008; Chen and Dorn, 2013; Sugiura et al., 2013; Gong et al., 2015; Leal et al., 2016; Naon et al., 2016, 2017; Boutant et al., 2017; Filadi et al., 2017; Basso et al., 2018; McLelland et al., 2018; Hernández-Alvarez et al., 2019). While MFN2 CMT mutants reduce ER-mitochondria contacts (Bernard-Marissal et al., 2019; Larrea et al., 2019), it is unclear whether these changes affect mitochondrial mobility.
How alterations in mitochondrial motility impact mitochondrial function, particularly in the context of CMT, remains poorly understood. Despite clear defects in mitochondrial mobility, some studies have concluded CMT mutations do not alter readouts of mitochondrial OXPHOS function such as mitochondrial membrane potential, oxygen consumption, and ATP production, or impair cellular calcium levels which mitochondria are involved in controlling (Baloh et al., 2007; Larrea et al., 2019). However, other studies have demonstrated that CMT mutations cause defects in these readouts (Loiseau et al., 2007; Guillet et al., 2010; Barneo-Munoz et al., 2015; Saporta et al., 2015; Rocha et al., 2018; Almutawa et al., 2019; Bernard-Marissal et al., 2019). And, another study demonstrated that bioenergetic efficiency and viability in a fly model can be rescued with only minor alterations in mitochondrial distribution (Trevisan et al., 2018). These discrepancies may be at least partially explained by differences in the model systems and experimental conditions used. There are now a wide variety of tools to study CMT including mouse and fly (Drosophila melanogaster) genetic models and iPSC-derived motor neurons (Saporta et al., 2015; Yamaguchi and Takashima, 2018; Juneja et al., 2019).
There is also clear evidence for a role of organelle-organelle contacts affecting mitochondrial mobility in CMT caused by mutations in the endo-lysosomal protein RAB7A. Wong et al. demonstrated reduced mitochondrial mobility due to prolonged inter-mitochondrial contacts in HeLa cells expressing CMT-mutant MFN2, RAB7A, or TRPV4 (Transient Receptor Potential Cation Channel Subfamily V Member 4) (Wong et al., 2019). RAB7A CMT mutations also increase tethering between mitochondria and endolysosomes, leading to changes in mitochondrial morphology and reduced mitochondrial mobility (Wong et al., 2018, 2019; Cioni et al., 2019). There is also evidence pointing towards an interaction between RAB7A and MFN2 (Zhao T. et al., 2012). Together, these findings suggest that interpretations of RAB7A mutations causing CMT based solely on defects in its endo-lysosomal function may be too simplistic. In the same vein, a recent study found that CMT-causing GDAP1 (Ganglioside Induced Differentiation Associated Protein 1) mutations result in defective mitochondria-lysosome contacts (Cantarero et al., 2020). That mitochondria-organelle contacts can affect mitochondrial mobility and dynamics highlights the limitations of evaluating protein and organelle dysfunction in isolation.
Recently, a screen for RAB7A binding partners found that another CMT protein, INF2 (Inverted Formin 2), is one of several actin-binding candidate interaction partners for RAB7A (Pan et al., 2020). This is particularly relevant to our discussion on CMT, inter-organelle contacts, and mitochondrial mobility for multiple reasons. First, a splice isoform of INF2 is tail-anchored to the ER. Second, dominant active mutations in ER-anchored INF2 that mimic INF2 CMT mutations have been shown to increase actin-dependent mitochondrial fragmentation and decrease mitochondrial mobility (Korobova et al., 2013; Chakrabarti et al., 2018). Together, these data point towards an important role in ER-mitochondria inter-organelle contacts in somehow regulating mitochondrial mobility via the actin cytoskeleton. That INF2 also potentially interacts with RAB7A suggests that mitochondria, endo-lysosomes, and ER all contact one another via CMT-associated proteins.
All INF2 CMT mutations are predicted or have been shown to increase actin assembly (Bayraktar et al., 2020). While some actin-binding motor proteins likely facilitate microtubule-independent mitochondrial transport, numerous studies have shown that long-range microtubule-based mobility of mitochondria is antagonized by actin and actin-binding motor proteins (Chada and Hollenbeck, 2004; Quintero et al., 2009; Pathak et al., 2010; Venkatesh et al., 2019; Cardanho-Ramos et al., 2020). Thus, while the effects of INF2 CMT mutations have yet to be studied in neurons, it is reasonable to expect that INF2 CMT mutations will cause an actin-dependent decrease in mitochondrial mobility in axons. Furthermore, since the ER regularly contacts many other organelles, and even appears to drive actin-assembly at ER-organelle contact sites (Korobova et al., 2013, 2014; Manor et al., 2015; Chakrabarti et al., 2018; Yang and Svitkina, 2019; Schiavon et al., 2020), it is quite possible INF2 CMT mutations cause aberrant actin assembly on other organelles, reducing their mobility as well (Figure 1).
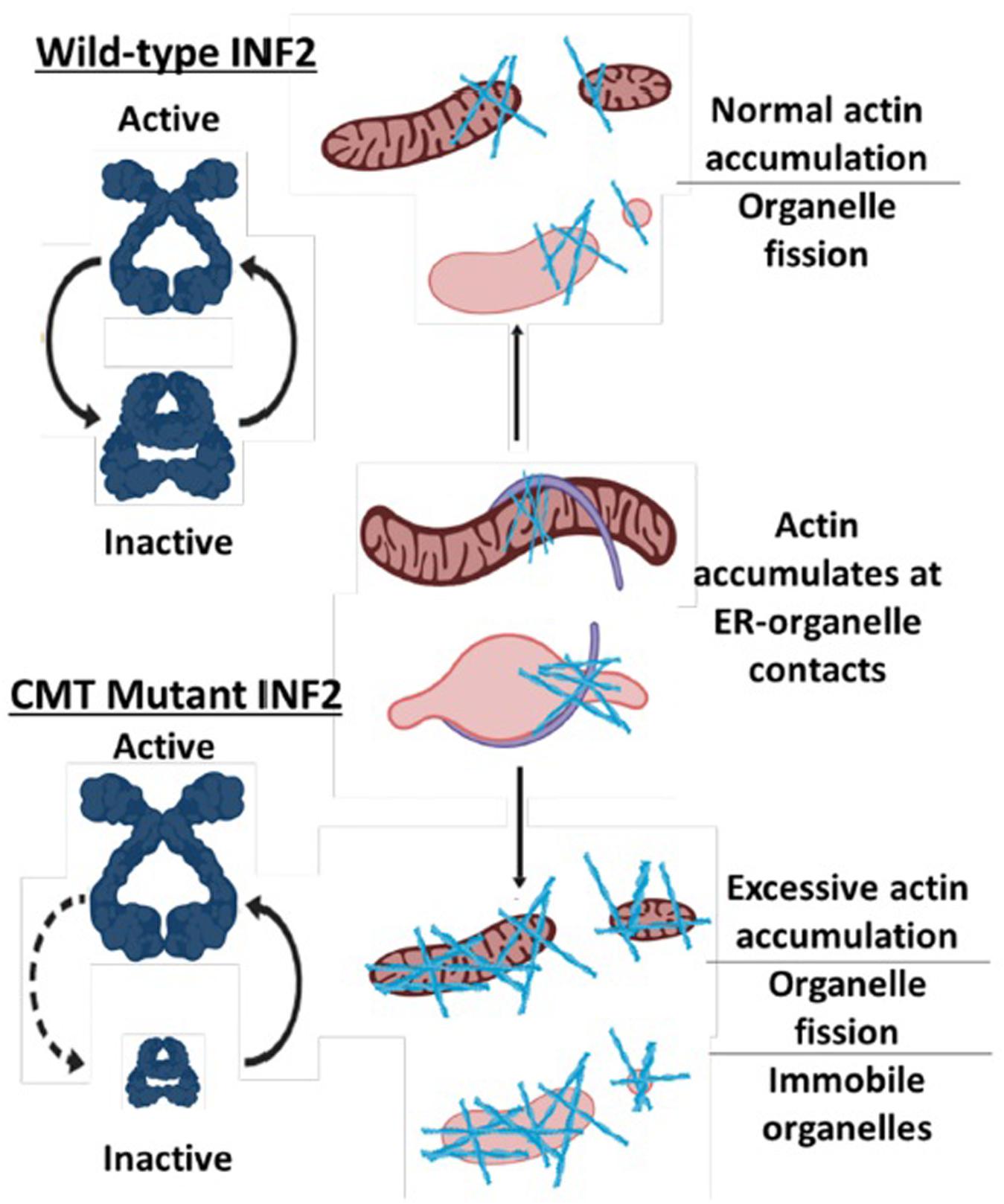
Figure 1. Model for how CMT mutations in INF2 reduce mitochondrial mobility. Normal INF2 is autoinhibited as indicated by open conformation compared to the closed conformation and thus has regulated actin-assembly activity. CMT mutations in INF2 reduce autoinhibition, resulting in excessive actin assembly on mitochondria and potentially other organelles, which in turn reduces their mobility.
Together with the newly uncovered role for RAB7A in (indirectly) modulating actin assembly (Pan et al., 2020), these observations point towards a role for multiple CMT mutations causing aberrant organelle-organelle and organelle-actin contacts, all of which cause decreased mitochondrial mobility. Whether some (or all) CMT mutations also cause decreased mobility of other organelles remains an important open question.
The focus of the role of mitochondria in CMT has been primarily on MFN2 and GDAP1, and to a lesser extent on associated motor proteins (KIF1B – Kinesin Family Member 1B, KIF5A – Kinesin Family Member 5A, DYNC1H1 – Dynein Cytoplasmic 1 Heavy Chain 1, DCTN2 – Dynactin Subunit 2) and some cytoskeletal proteins (NEFL – Neurofilament Light). Here, we have highlighted INF2 and RAB7A as CMT-associated proteins likely involved in mitochondrial mobility and dynamics. However, we propose that the proteins mutated in CMT that play roles in mitochondrial function, dynamics and mobility likely extend well beyond just these two (see Table 1 for a full list).
Why Do Mobility Defects Usually Only Affect Peripheral Neurons in CMT Patients?
Hopefully, we have provided a convincing argument that many CMT mutations likely reduce mitochondrial mobility. Given the extreme lengths of peripheral axons, it is tempting to conclude that a reduction in mobility due to CMT mutations simply affects longer axons more severely (the “impaired mobility model” of CMT). One can easily reconcile two key features of CMT using the impaired mobility model: The progressive nature of the disease: This suggests that dysfunction must accumulate over time. One can imagine this more severely affects longer axons, due to reduced turnover of damaged mitochondria resulting from reduced mitochondrial mobility. Interestingly, one could imagine that reduced mobility of other organelles associated with turnover (e.g., lysosomes) could also cause increased accumulation of damaged mitochondria in longer axons. The longest peripheral axons (i.e., the feet) progressively degenerate prior to the next-longest axons (i.e., the hands): This further supports the impaired mobility model, wherein damage accumulates first in the longest axons due to the more demanding, “longer commute” resulting in faster accumulation of damaged mitochondria.
Unfortunately, while this model is compelling, it appears to be overly simplistic. The weakness in relying on mobility alone as an explanation can best be highlighted by comparing the lengths of different axons both within and between species. For example, the longest axon in mice is approximately 2 cm, whereas in humans the longest axon is ∼60 times longer. Just as striking, some unaffected axons in the human CNS may be longer than the mouse’s longest axon. The very same mutation in humans and mice can cause CMT, yet no defects are found in the brains of human CMT patients. Meanwhile, the motor proteins, cytoskeletal tracks, and mitochondria of mice and humans are all roughly the same size, and all possess roughly the same biophysical properties (e.g., velocity, force generation, etc.) when transporting their organelles across long distances (Figure 2).
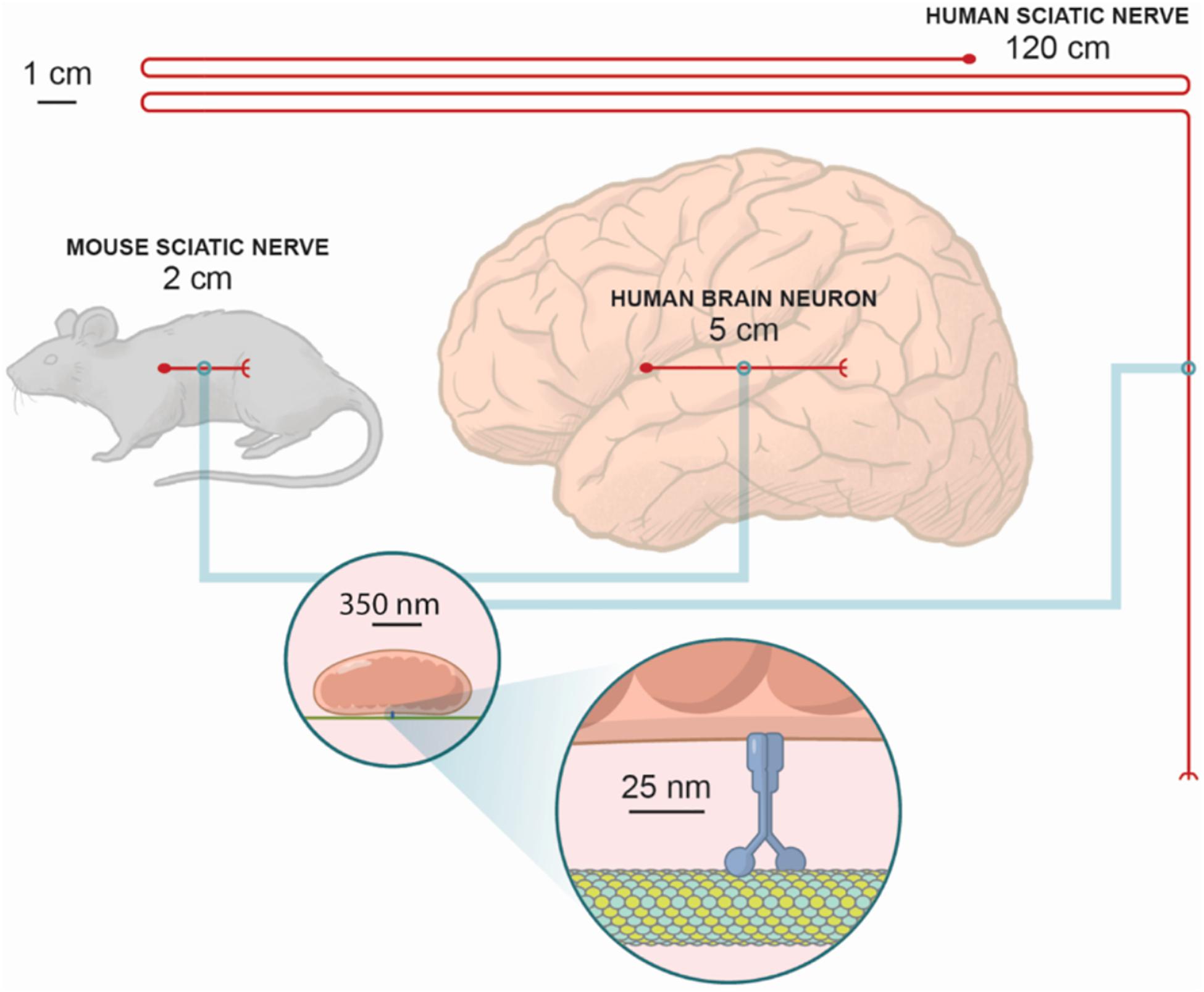
Figure 2. The relative scales of axons in humans versus mice. While microtubules, motor proteins, and mitochondria have nearly identical sizes in mice and men, the length of the axons can be very different. At the same time, axons in the mouse sciatic nerve are not as long as some of the longer axons within the human brain. For example, it is known in the macaque there are direct connections from the frontal eye fields in the anterior bank of the arcuate sulcus to the primary visual cortex, which in the macaque is ∼5 cm, and probably as long as 12.5 cm in humans. Notably, for most CMT patients the pathology is often constrained to only peripheral neurons. Since the same mutations can cause CMT in mice and men, the distances mitochondria must travel can only provide a partial explanation for the physiopathology of these mutations.
Any analysis of mobility must consider not just distance but also time. Most laboratory mice only live for ∼2 years, while human CMT patients may not even experience symptoms until adolescence or adulthood. Thus, there is clearly a “missing variable” that underlies differences in lifespan and disease susceptibility between species (e.g., differences in metabolism or oxidative stress). Thus, CMT may serve as a “model disease” to better understand age-related neurodegeneration. It is well established mitochondria play myriad roles at the pre-synapse, including ATP production, intra- and intercellular signaling (e.g., calcium signaling and signaling via reactive oxygen species), and the biosynthesis of signaling molecules (e.g., lipids, hormones, and neurotransmitter intermediates) (Devine and Kittler, 2018). Perhaps perturbed transport of mitochondria to the pre-synapse of peripheral neurons in CMT provides an opportunity to better understand other neurodegenerative disorders associated with defective presynaptic mitochondria, including Alzheimer’s, ALS, Parkinson’s, Friedreich’s Ataxia, and Hereditary Spastic Paraplegia (Devine and Kittler, 2018).
But even considering the impaired mobility model for CMT within a single organism has some issues. It is difficult to imagine how reductions in mobility as high as 100% (Baloh et al., 2007; Rocha et al., 2018) could have a severe effect only on the longest axons, but not other axons that, while shorter than the peripheral neurons, are still very long compared to the ∼25 nm step size of a motor protein.
When considering these conundrums, it is helpful to consider alternative mechanisms for replenishing mitochondria in neurons, many of which are reviewed elsewhere (Misgeld and Schwarz, 2017; Yu and Pekkurnaz, 2018). Briefly, mitochondrial rejuvenation is speculated to be at least partially mediated via local translation in the axon. Interestingly, multiple CMT mutations affect local translation machinery (Table 1). More recent work showed that mitochondria serve as a stable compartment for mediating biogenesis by serving as an energy source for synaptic translation (Rangaraju et al., 2019). This raises a chicken vs. egg question: Does reduced mitochondrial mobility impair local translation needed for synaptic and therefore neuronal health and maintenance? Or does impaired local translation lead to dysfunctional mitochondria that cannot be replaced without sufficient mobility? That CMT is caused by mutations disrupting both mobility and local translation indicates these two processes have a unique relationship in long axons.
Conclusion and Open Questions
One open question is how the overall distribution of mitochondria is altered in CMT neurons, and how this relates to axonal maintenance. A recent study showed mitochondria tend to distribute along the length of axons with regular spacing, and that inter-mitochondrial feedback mediates their positioning and movement (Matsumoto et al., 2020). Is this feedback-based positioning altered in CMT? Do mutations affecting mobility result in CMT via a “domino effect” caused by defects in relatively local repositioning between axonal mitochondria, which then cascades with increasing defects as a function of increasing axonal length? How much longer does it take mitochondria in CMT patients to traverse the entire length of an axon? Defective mitophagy has been implicated in other neurodegenerative disorders and some studies have linked CMT to alterations in autophagy (Colecchia et al., 2018; Gautam et al., 2019). Is there a reduction in the turnover rate of mitochondria in CMT patients? Mitochondria are increasingly being implicated as important players in adaptive and innate immune responses and inflammatory pathology, including neurodegeneration (West, 2017; Newman and Shadel, 2018). Could “mitoflammation” contribute to the pathophysiology of CMT? How do any and all of these factors affect mitochondria at the pre-synapse of CMT peripheral neurons, likely the most important subpopulation of mitochondria in these cells? These are surprisingly open questions we expect to be addressed in the coming years using animal and cell models of CMT.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The Waitt Advanced Biophotonics Center was funded by the Waitt Foundation and Core Grant application National Cancer Institute (NCI) Cancer Center Support Grant (CCSG; CA014195). GS is the Audrey Geisel Chair in Biomedical Science and supported by NIH R01 AR069876. UM is a Chan Zuckerberg Initiative Imaging Scientists and supported by NIH R21 DC018237. CS was supported by NIH F32 GM137580.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Timothy Shutt for critical feedback on the original version of the manuscript. We are grateful to Amy Cao (Salk Institute for Biological Studies) for help with the cartoon diagram in Figure 2.
References
Albulym, O. M., Kennerson, M. L., Harms, M. B., Drew, A. P., Siddell, A. H., Auer-Grumbach, M., et al. (2016). MORC2 mutations cause axonal Charcot-Marie-Tooth disease with pyramidal signs. Ann. Neurol. 79, 419–427. doi: 10.1002/ana.24575
Almutawa, W., Smith, C., Sabouny, R., Smit, R. B., Zhao, T., Wong, R., et al. (2019). The R941L mutation in MYH14 disrupts mitochondrial fission and associates with peripheral neuropathy. EBioMedicine 45, 379–392. doi: 10.1016/j.ebiom.2019.06.018
Ammar, N., Nelis, E., Merlini, L., Barisić, N., Amouri, R., Ceuterick, C., et al. (2003). Identification of novel GDAP1 mutations causing autosomal recessive Charcot-Marie-Tooth disease. Neuromuscul. Disord. 13, 720–728. doi: 10.1016/s0960-8966(03)00093-2
Amornvit, J., Yalvac, M. E., Chen, L., and Sahenk, Z. (2017). A novel p.T139M mutation in HSPB1 highlighting the phenotypic spectrum in a family. Brain Behav. 7:e00774. doi: 10.1002/brb3.774
Ando, M., Hashiguchi, A., Okamoto, Y., Yoshimura, A., Hiramatsu, Y., Yuan, J., et al. (2017). Clinical and genetic diversities of Charcot-Marie-Tooth disease with MFN2 mutations in a large case study. J. Peripher. Nerv. Syst. 22, 191–199. doi: 10.1111/jns.12228
Auer-Grumbach, M., Fischer, C., Papiæ, L., John, E., Plecko, B., Bittner, R. E., et al. (2008). Two novel mutations in the GDAP1 and PRX genes in early onset Charcot-Marie-Tooth syndrome. Neuropediatrics 39, 33–38. doi: 10.1055/s-2008-1077085
Auer-Grumbach, M., Toegel, S., Schabhüttl, M., Weinmann, D., Chiari, C., Bennett, D. L. H., et al. (2016). Rare variants in MME, Encoding metalloprotease neprilysin, are linked to late-onset autosomal-dominant axonal polyneuropathies. Am. J. Hum. Genet. 99, 607–623. doi: 10.1016/j.ajhg.2016.07.008
Auranen, M., Ylikallio, E., Toppila, J., Somer, M., Kiuru-Enari, S., and Tyynismaa, H. (2013). Dominant GDAP1 founder mutation is a common cause of axonal Charcot-Marie-Tooth disease in Finland. Neurogenetics 14, 123–132. doi: 10.1007/s10048-013-0358-9
Avila, R. L., D’Antonio, M., Bachi, A., Inouye, H., Feltri, M. L., Wrabetz, L., et al. (2010). P0 (protein zero) mutation S34C underlies instability of internodal myelin in S63C mice. J. Biol. Chem. 285, 42001–42012. doi: 10.1074/jbc.M110.166967
Baets, J., De Jonghe, P., and Timmerman, V. (2014). Recent advances in Charcot-Marie-Tooth disease. Curr. Opin. Neurol. 27, 532–540. doi: 10.1097/wco.0000000000000131
Baloh, R. H., Schmidt, R. E., Pestronk, A., and Milbrandt, J. (2007). Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J. Neurosci. 27, 422–430. doi: 10.1523/JNEUROSCI.4798-06.2007
Bannerman, P., Burns, T., Xu, J., Miers, L., and Pleasure, D. (2016). Mice hemizygous for a pathogenic mitofusin-2 allele exhibit hind limb/foot gait deficits and phenotypic perturbations in nerve and muscle. PLoS One 11:e0167573. doi: 10.1371/journal.pone.0167573
Bansagi, B., Antoniadi, T., Burton-Jones, S., Murphy, S. M., McHugh, J., Alexander, M., et al. (2015). Genotype/phenotype correlations in AARS-related neuropathy in a cohort of patients from the United Kingdom and Ireland. J. Neurol. 262, 1899–1908. doi: 10.1007/s00415-015-7778-4
Baránková, L., Vyhnálková, E., Züchner, S., Mazanec, R., Sakmaryová, I., Vondrácek, P., et al. (2007). GDAP1 mutations in Czech families with early-onset CMT. Neuromuscul. Disord. 17, 482–489. doi: 10.1016/j.nmd.2007.02.010
Barneo-Munoz, M., Juarez, P., Civera-Tregon, A., Yndriago, L., Pla-Martin, D., Zenker, J., et al. (2015). Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of charcot-marie-tooth neuropathy. PLoS Genet. 11:e1005115. doi: 10.1371/journal.pgen.1005115
Barreto, L. C., Oliveira, F. S., Nunes, P. S., de França Costa, I. M., Garcez, C. A., Goes, G. M., et al. (2016). Epidemiologic study of charcot-marie-tooth disease: a systematic review. Neuroepidemiology 46, 157–165. doi: 10.1159/000443706
Basso, V., Marchesan, E., Peggion, C., Chakraborty, J., von Stockum, S., Giacomello, M., et al. (2018). Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 138, 43–56. doi: 10.1016/j.phrs.2018.09.006
Baxter, R. V., Ben Othmane, K., Rochelle, J. M., Stajich, J. E., Hulette, C., Dew-Knight, S., et al. (2002). Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 30, 21–22. doi: 10.1038/ng796
Bayraktar, S., Nehrig, J., Menis, E., Karli, K., Janning, A., Struk, T., et al. (2020). A Deregulated stress response underlies distinct INF2-Associated disease profiles. J. Am. Soc. Nephrol. 31, 1296–1313. doi: 10.1681/asn.2019111174
Berciano, J., Peeters, K., García, A., López-Alburquerque, T., Gallardo, E., Hernández-Fabián, A., et al. (2016). NEFL N98S mutation: another cause of dominant intermediate Charcot-Marie-Tooth disease with heterogeneous early-onset phenotype. J. Neurol. 263, 361–369. doi: 10.1007/s00415-015-7985-z
Beręsewicz, M., Boratyńska-Jasińska, A., Charzewski, Ł, Kawalec, M., Kabzińska, D., Kochański, A., et al. (2017). The Effect of a Novel c.820C>T (Arg274Trp) mutation in the mitofusin 2 gene on fibroblast metabolism and clinical manifestation in a patient. PLoS One 12:e0169999. doi: 10.1371/journal.pone.0169999
Bergamin, G., Boaretto, F., Briani, C., Pegoraro, E., Cacciavillani, M., Martinuzzi, A., et al. (2014). Mutation analysis of MFN2, GJB1, MPZ and PMP22 in Italian patients with axonal Charcot-Marie-Tooth disease. Neuromolecular. Med. 16, 540–550. doi: 10.1007/s12017-014-8307-9
Bernard-Marissal, N., van Hameren, G., Juneja, M., Pellegrino, C., Louhivuori, L., Bartesaghi, L., et al. (2019). Altered interplay between endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type 2A neuropathy. Proc. Natl. Acad. Sc.i U.S.A. 116, 2328–2337. doi: 10.1073/pnas.1810932116
Bhagavati, S., Maccabee, P. J., and Xu, W. (2009). The neurofilament light chain gene (NEFL) mutation Pro22Ser can be associated with mixed axonal and demyelinating neuropathy. J. Clin. Neurosci. 16, 830–831. doi: 10.1016/j.jocn.2008.08.030
Bian, X., Lin, P., Li, J., Long, F., Duan, R., Yuan, Q., et al. (2018). Whole-genome linkage analysis with whole-exome sequencing identifies a novel frameshift variant in NEFH in a Chinese family with Charcot-Marie-Tooth 2: a novel variant in NEFH for Charcot-Marie-Tooth 2. Neurodegener. Dis. 18, 74–83. doi: 10.1159/000487754
Bienfait, H. M., Faber, C. G., Baas, F., Gabreëls-Festen, A. A., Koelman, J. H., Hoogendijk, J. E., et al. (2006). Late onset axonal Charcot-Marie-Tooth phenotype caused by a novel myelin protein zero mutation. J. Neurol. Neurosurg. Psychiatry 77, 534–537. doi: 10.1136/jnnp.2005.073437
Bissar-Tadmouri, N., Gulsen-Parman, Y., Latour, P., Deymeer, F., Serdaroglu, P., Vandenberghe, A., et al. (1999). Two novel mutations in the MPZ gene coding region in Charcot-Marie-Tooth type 1 patients of Turkish origin: S54P, [I30del; GVYI29ins]. Hum. Mutat. 14:449. doi: 10.1002/(sici)1098-1004(199911)14:5<449::Aid-humu17<3.0.Co;2-h
Bissar-Tadmouri, N., Nelis, E., Züchner, S., Parman, Y., Deymeer, F., Serdaroglu, P., et al. (2004). Absence of KIF1B mutation in a large Turkish CMT2A family suggests involvement of a second gene. Neurology 62, 1522–1525. doi: 10.1212/01.wnl.0000123253.57555.3a
Bitoun, M., Stojkovic, T., Prudhon, B., Maurage, C. A., Latour, P., Vermersch, P., et al. (2008). A novel mutation in the dynamin 2 gene in a Charcot-Marie-Tooth type 2 patient: clinical and pathological findings. Neuromuscul. Disord. 18, 334–338. doi: 10.1016/j.nmd.2008.01.005
Blanquet-Grossard, F., Pham-Dinh, D., Dautigny, A., Latour, P., Bonnebouche, C., Diraison, P., et al. (1996). Charcot-Marie-Tooth type 1B neuropathy: a mutation at the single glycosylation site in the major peripheral myelin glycoprotein Po. Hum. Mutat. 8, 185–186. doi: 10.1002/(sici)1098-100419968:2<185::Aid-humu13<3.0.Co;2-z
Boutant, M., Kulkarni, S. S., Joffraud, M., Ratajczak, J., Valera-Alberni, M., Combe, R., et al. (2017). Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 36, 1543–1558. doi: 10.15252/embj.201694914
Boyer, O., Benoit, G., Gribouval, O., Nevo, F., Tete, M. J., Dantal, J., et al. (2011a). Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 22, 239–245. doi: 10.1681/ASN.2010050518
Boyer, O., Nevo, F., Plaisier, E., Funalot, B., Gribouval, O., Benoit, G., et al. (2011b). INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N. Engl. J. Med. 365, 2377–2388. doi: 10.1056/NEJMoa1109122
Braathen, G. J., Høyer, H., Busk, ØL., Tveten, K., Skjelbred, C. F., and Russell, M. B. (2016). Variants in the genes DCTN2, DNAH10, LRIG3, and MYO1A are associated with intermediate Charcot-Marie-Tooth disease in a Norwegian family. Acta Neurol. Scand. 134, 67–75. doi: 10.1111/ane.12515
Braathen, G. J., Sand, J. C., Lobato, A., Hoyer, H., and Russell, M. B. (2010). MFN2 point mutations occur in 3.4% of Charcot-Marie-Tooth families. An investigation of 232 Norwegian CMT families. BMC Med. Genet. 11:48. doi: 10.1186/1471-2350-11-48
Brozková, D., Mazanec, R., Haberlová, J., Sakmaryová, I., and Seeman, P. (2010). Clinical and in silico evidence for and against pathogenicity of 11 new mutations in the MPZ gene. Clin. Genet. 78, 81–87. doi: 10.1111/j.1399-0004.2010.01423.x
Brožková, D., Posádka, J., Laššuthová, P., Mazanec, R., Haberlová, J., Sišková, D., et al. (2013). Spectrum and frequencies of mutations in the MFN2 gene and its phenotypical expression in Czech hereditary motor and sensory neuropathy type II patients. Mol. Med. Rep. 8, 1779–1784. doi: 10.3892/mmr.2013.1730
Calvo, J., Funalot, B., Ouvrier, R. A., Lazaro, L., Toutain, A., De Mas, P., et al. (2009). Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch. Neurol. 66, 1511–1516. doi: 10.1001/archneurol.2009.284
Cantarero, L., Juárez-Escoto, E., Civera-Tregón, A., Rodríguez-Sanz, M., Roldán, M., Benítez, R., et al. (2020). Mitochondria–Lysosome membrane contacts are defective in GDAP1-Related Charcot–Marie–Tooth disease. Hum. Mol. Genet. ddaa243. doi: 10.1093/hmg/ddaa243
Cardanho-Ramos, C., Faria-Pereira, A., and Morais, V. A. (2020). Orchestrating mitochondria in neurons: cytoskeleton as the conductor. Cytoskeleton 77, 65–75. doi: 10.1002/cm.21585
Caridi, G., Lugani, F., Dagnino, M., Gigante, M., Iolascon, A., Falco, M., et al. (2014). Novel INF2 mutations in an Italian cohort of patients with focal segmental glomerulosclerosis, renal failure and Charcot-Marie-Tooth neuropathy. Nephrol. Dial. Transplant.z 29(Suppl. 4), iv80–iv86. doi: 10.1093/ndt/gfu071
Cartoni, R., and Martinou, J. C. (2009). Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp. Neurol. 218, 268–273. doi: 10.1016/j.expneurol.2009.05.003
Cassereau, J., Chevrollier, A., Bonneau, D., Verny, C., Procaccio, V., Reynier, P., et al. (2011). A locus-specific database for mutations in GDAP1 allows analysis of genotype-phenotype correlations in Charcot-Marie-Tooth diseases type 4A and 2K. Orphanet. J. Rare Dis. 6:87. doi: 10.1186/1750-1172-6-87
Chada, S. R., and Hollenbeck, P. J. (2004). Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr. Biol. 14, 1272–1276. doi: 10.1016/j.cub.2004.07.027
Chakrabarti, R., Ji, W. K., Stan, R. V., de Juan Sanz, J., Ryan, T. A., and Higgs, H. N. (2018). INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 217, 251–268. doi: 10.1083/jcb.201709111
Chaouch, M., Allal, Y., De Sandre-Giovannoli, A., Vallat, J. M., Amer-el-Khedoud, A., Kassouri, N., et al. (2003). The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul. Disord. 13, 60–67. doi: 10.1016/s0960-8966(02)00196-7
Chapman, A. L., Bennett, E. J., Ramesh, T. M., De Vos, K. J., and Grierson, A. J. (2013). Axonal transport defects in a mitofusin 2 loss of function model of Charcot-Marie-Tooth Disease in Zebrafish. PLoS One 8:e67276. doi: 10.1371/journal.pone.0067276
Chaudhry, R., Kidambi, A., Brewer, M. H., Antonellis, A., Mathews, K., Nicholson, G., et al. (2013). Re-analysis of an original CMTX3 family using exome sequencing identifies a known BSCL2 mutation. Muscle Nerve 47, 922–924. doi: 10.1002/mus.23743
Chavada, G., Rao, D. G., Martindale, J., and Hadjivassiliou, M. (2012). A novel MPZ gene mutation in exon 2 causing late-onset demyelinating Charcot-Marie-Tooth disease. J. Clin. Neuromuscul. Dis. 13, 206–208. doi: 10.1097/CND.0b013e3182461a83
Chen, Y., and Dorn, G. W. II (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Cho, H. J., Sung, D. H., Kim, B. J., and Ki, C. S. (2007). Mitochondrial GTPase mitofusin 2 mutations in Korean patients with Charcot-Marie-Tooth neuropathy type 2. Clin. Genet. 71, 267–272. doi: 10.1111/j.1399-0004.2007.00763.x
Choi, B. O., Kim, S. B., Kanwal, S., Hyun, Y. S., Park, S. W., Koo, H., et al. (2011). MPZ mutation in an early-onset Charcot-Marie-Tooth disease type 1B family by genome-wide linkage analysis. Int. J. Mol. Med. 28, 389–396. doi: 10.3892/ijmm.2011.678
Choi, B. O., Nakhro, K., Park, H. J., Hyun, Y. S., Lee, J. H., Kanwal, S., et al. (2015). A cohort study of MFN2 mutations and phenotypic spectrums in Charcot-Marie-Tooth disease 2A patients. Clin. Genet. 87, 594–598. doi: 10.1111/cge.12432
Choi, Y. R., Hong, Y. B., Jung, S. C., Lee, J. H., Kim, Y. J., Park, H. J., et al. (2015). A novel homozygous MPV17 mutation in two families with axonal sensorimotor polyneuropathy. BMC Neurol. 15:179. doi: 10.1186/s12883-015-0430-1
Chung, K. W., Kim, S. B., Cho, S. Y., Hwang, S. J., Park, S. W., Kang, S. H., et al. (2008). Distal hereditary motor neuropathy in Korean patients with a small heat shock protein 27 mutation. Exp. Mol. Med. 40, 304–312. doi: 10.3858/emm.2008.40.3.304
Chung, K. W., Kim, S. B., Park, K. D., Choi, K. G., Lee, J. H., Eun, H. W., et al. (2006). Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain 129(Pt 8), 2103–2118. doi: 10.1093/brain/awl174
Cioni, J. M., Lin, J. Q., Holtermann, A. V., Koppers, M., Jakobs, M. A. H., Azizi, A., et al. (2019). Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56.e15–72.e15. doi: 10.1016/j.cell.2018.11.030
Codron, P., Chevrollier, A., Kane, M. S., Echaniz-Laguna, A., Latour, P., Reynier, P., et al. (2016). Increased mitochondrial fusion in a autosomal recessive CMT2A family with mitochondrial GTPase mitofusin 2 mutations. J. Peripher. Nerv. Syst. 21, 365–369. doi: 10.1111/jns.12192
Colecchia, D., Stasi, M., Leonardi, M., Manganelli, F., Nolano, M., Veneziani, B. M., et al. (2018). Alterations of autophagy in the peripheral neuropathy Charcot-Marie-Tooth type 2B. Autophagy 14, 930–941. doi: 10.1080/15548627.2017.1388475
Cottenie, E., Kochanski, A., Jordanova, A., Bansagi, B., Zimon, M., Horga, A., et al. (2014). Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am. J. Hum. Genet. 95, 590–601. doi: 10.1016/j.ajhg.2014.10.002
Cuesta, A., Pedrola, L., Sevilla, T., García-Planells, J., Chumillas, M. J., Mayordomo, F., et al. (2002). The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 30, 22–25. doi: 10.1038/ng798
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Jonghe, P., Timmerman, V., Ceuterick, C., Nelis, E., De Vriendt, E., Löfgren, A., et al. (1999). The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain 122(Pt 2), 281–290. doi: 10.1093/brain/122.2.281
De Sandre-Giovannoli, A., Chaouch, M., Kozlov, S., Vallat, J. M., Tazir, M., Kassouri, N., et al. (2002). Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 70, 726–736. doi: 10.1086/339274
Del Bo, R., Moggio, M., Rango, M., Bonato, S., D’Angelo, M. G., Ghezzi, S., et al. (2008). Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 71, 1959–1966. doi: 10.1212/01.wnl.0000327095.32005.a4
Deng, H. X., Klein, C. J., Yan, J., Shi, Y., Wu, Y., Fecto, F., et al. (2010). Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat. Genet. 42, 165–169. doi: 10.1038/ng.509
Detmer, S. A., and Chan, D. C. (2007). Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 176, 405–414. doi: 10.1083/jcb.200611080
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nrn.2017.170
Di Maria, E., Gulli, R., Balestra, P., Cassandrini, D., Pigullo, S., Doria-Lamba, L., et al. (2004). A novel mutation of GDAP1 associated with Charcot-Marie-Tooth disease in three Italian families: evidence for a founder effect. J. Neurol. Neurosurg. Psychiatry 75, 1495–1498. doi: 10.1136/jnnp.2003.028100
Di Meglio, C., Bonello-Palot, N., Boulay, C., Milh, M., Ovaert, C., Levy, N., et al. (2016). Clinical and allelic heterogeneity in a pediatric cohort of 11 patients carrying MFN2 mutation. Brain Dev. 38, 498–506. doi: 10.1016/j.braindev.2015.11.006
Dohrn, M. F., Glöckle, N., Mulahasanovic, L., Heller, C., Mohr, J., Bauer, C., et al. (2017). Frequent genes in rare diseases: panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J. Neurochem. 143, 507–522. doi: 10.1111/jnc.14217
Echaniz-Laguna, A., and Latour, P. (2019). A cryptic splicing mutation in the INF2 gene causing Charcot-Marie-Tooth disease with minimal glomerular dysfunction. J. Peripher. Nerv. Syst. 24, 120–124. doi: 10.1111/jns.12308
Echaniz-Laguna, A., Nicot, A. S., Carré, S., Franques, J., Tranchant, C., Dondaine, N., et al. (2007). Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul. Disord. 17, 955–959. doi: 10.1016/j.nmd.2007.06.467
El Fissi, N., Rojo, M., Aouane, A., Karatas, E., Poliacikova, G., David, C., et al. (2018). Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT2A neuropathy. EMBO Rep. 19:e45241. doi: 10.15252/embr.201745241
Engelfried, K., Vorgerd, M., Hagedorn, M., Haas, G., Gilles, J., Epplen, J. T., et al. (2006). Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2). BMC Med. Genet. 7:53. doi: 10.1186/1471-2350-7-53
Fabrizi, G. M., Cavallaro, T., Angiari, C., Cabrini, I., Taioli, F., Malerba, G., et al. (2007). Charcot-Marie-Tooth disease type 2E, a disorder of the cytoskeleton. Brain 130(Pt 2), 394–403. doi: 10.1093/brain/awl284
Feely, S. M., Laura, M., Siskind, C. E., Sottile, S., Davis, M., Gibbons, V. S., et al. (2011). MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 76, 1690–1696. doi: 10.1212/WNL.0b013e31821a441e
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2017). On the role of Mitofusin 2 in endoplasmic reticulum-mitochondria tethering. Proc. Natl. Acad. Sci. U.S.A. 114, E2266–E2267. doi: 10.1073/pnas.1616040114
Finsterer, J., Fiorini, A. C., Scorza, C. A., and Scorza, F. A. (2018). CMT2 due to homozygous MFN2 variants is a multiorgan mitochondrial disorder. Eur. J. Paediatr. Neurol. 22, 889–891. doi: 10.1016/j.ejpn.2018.04.012
Fu, J., Ma, M., Pang, M., Yang, L., Li, G., Song, J., et al. (2019). [Analysis of a pedigree with autosomal dominant intermediate Charcot-Marie-Tooth disease type E and nephropathy]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 36, 918–921. doi: 10.3760/cma.j.issn.1003-9406.2019.09.016
Fu, J., and Yuan, Y. (2018). A novel homozygous nonsense mutation in NEFL causes autosomal recessive Charcot-Marie-Tooth disease. Neuromuscul. Disord. 28, 44–47. doi: 10.1016/j.nmd.2017.09.018
Fujisawa, M., Sano, Y., Omoto, M., Ogasawara, J. I., Koga, M., Takashima, H., et al. (2017). [Charcot-Marie-Tooth disease type 2 caused by homozygous MME gene mutation superimposed by chronic inflammatory demyelinating polyneuropathy]. Rinsho Shinkeigaku 57, 515–520. doi: 10.5692/clinicalneurol.cn-001036
Fusco, C., Ucchino, V., Barbon, G., Bonini, E., Mostacciuolo, M. L., Frattini, D., et al. (2011). The homozygous ganglioside-induced differentiation-associated protein 1 mutation c.373C > T causes a very early-onset neuropathy: case report and literature review. J. Child. Neurol. 26, 49–57. doi: 10.1177/0883073810373142
Gallardo, E., García, A., Ramón, C., Maraví, E., Infante, J., Gastón, I., et al. (2009). Charcot–Marie–Tooth disease type 2J with MPZ Thr124Met mutation: clinico-electrophysiological and MRI study of a family. J. Neurol. 256, 2061–2071. doi: 10.1007/s00415-009-5251-y
Gautam, M., Xie, E. F., Kocak, N., and Ozdinler, P. H. (2019). Mitoautophagy: a unique self-destructive path mitochondria of upper motor neurons With TDP-43 pathology take. very early in ALS. Front. Cell Neurosci. 13:489. doi: 10.3389/fncel.2019.00489
Gess, B., Auer-Grumbach, M., Schirmacher, A., Strom, T., Zitzelsberger, M., Rudnik-Schöneborn, S., et al. (2014). HSJ1-related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology 83, 1726–1732. doi: 10.1212/wnl.0000000000000966
Gong, G., Song, M., Csordas, G., Kelly, D. P., Matkovich, S. J., and Dorn, G. W. II (2015). Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350:aad2459. doi: 10.1126/science.aad2459
Gonzalez, M. A., Feely, S. M., Speziani, F., Strickland, A. V., Danzi, M., Bacon, C., et al. (2014). A novel mutation in VCP causes Charcot-Marie-Tooth Type 2 disease. Brain 137(Pt 11), 2897–2902. doi: 10.1093/brain/awu224
González-Jamett, A. M., Haro-Acuña, V., Momboisse, F., Caviedes, P., Bevilacqua, J. A., and Cárdenas, A. M. (2014). Dynamin-2 in nervous system disorders. J. Neurochem. 128, 210–223. doi: 10.1111/jnc.12455
Guernsey, D. L., Jiang, H., Bedard, K., Evans, S. C., Ferguson, M., Matsuoka, M., et al. (2010). Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet. 6:e1001081. doi: 10.1371/journal.pgen.1001081
Guillet, V., Gueguen, N., Verny, C., Ferre, M., Homedan, C., Loiseau, D., et al. (2010). Adenine nucleotide translocase is involved in a mitochondrial coupling defect in MFN2-related Charcot-Marie-Tooth type 2A disease. Neurogenetics 11, 127–133. doi: 10.1007/s10048-009-0207-z
Haberlová, J., Mazanec, R., Ridzoň, P., Baránková, L., Nürnberg, G., Nürnberg, P., et al. (2011). Phenotypic variability in a large Czech family with a dynamin 2-associated Charcot-Marie-Tooth neuropathy. J. Neurogenet. 25, 182–188. doi: 10.3109/01677063.2011.627484
Hamadouche, T., Poitelon, Y., Genin, E., Chaouch, M., Tazir, M., Kassouri, N., et al. (2008). Founder effect and estimation of the age of the c.892C>T (p.Arg298Cys) mutation in LMNA associated to Charcot-Marie-Tooth subtype CMT2B1 in families from North Western Africa. Ann. Hum. Genet. 72(Pt 5), 590–597. doi: 10.1111/j.1469-1809.2008.00456.x
Hamaguchi, A., Ishida, C., Iwasa, K., Abe, A., and Yamada, M. (2010). Charcot-Marie-Tooth disease type 2D with a novel glycyl-tRNA synthetase gene (GARS) mutation. J. Neurol. 257, 1202–1204. doi: 10.1007/s00415-010-5491-x
Hayasaka, K., Himoro, M., Sato, W., Takada, G., Uyemura, K., Shimizu, N., et al. (1993). Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat. Genet. 5, 31–34. doi: 10.1038/ng0993-31
He, J., Guo, L., Xu, G., Xu, L., Lin, S., Chen, W., et al. (2018). Clinical and genetic investigation in Chinese patients with demyelinating Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 23, 216–226. doi: 10.1111/jns.12277
Hernández-Alvarez, M. I., Sebastián, D., Vives, S., Ivanova, S., Bartoccioni, P., Kakimoto, P., et al. (2019). Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell 177, 881.e17–895.e17. doi: 10.1016/j.cell.2019.04.010
Ho, C. C., Tai, S. M., Lee, E. C., Mak, T. S., Liu, T. K., Tang, V. W., et al. (2017). Rapid identification of pathogenic variants in two cases of Charcot-Marie-Tooth disease by gene-panel sequencing. Int. J. Mol. Sci. 18:770. doi: 10.3390/ijms18040770
Hong, Y. B., Kang, J., Kim, J. H., Lee, J., Kwak, G., Hyun, Y. S., et al. (2016). DGAT2 mutation in a family with autosomal-dominant early-onset axonal Charcot-Marie-Tooth Disease. Hum. Mutat. 37, 473–480. doi: 10.1002/humu.22959
Horga, A., Laurà, M., Jaunmuktane, Z., Jerath, N. U., Gonzalez, M. A., Polke, J. M., et al. (2017). Genetic and clinical characteristics of NEFL-related Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 88, 575–585. doi: 10.1136/jnnp-2016-315077
Houlden, H., Laura, M., Wavrant-De Vrièze, F., Blake, J., Wood, N., and Reilly, M. M. (2008). Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology 71, 1660–1668. doi: 10.1212/01.wnl.0000319696.14225.67
Høyer, H., Braathen, G. J., Busk, ØL., Holla, ØL., Svendsen, M., Hilmarsen, H. T., et al. (2014). Genetic diagnosis of Charcot-Marie-Tooth disease in a population by next-generation sequencing. Biomed. Res. Int. 2014:210401. doi: 10.1155/2014/210401
Høyer, H., Braathen, G. J., Eek, A. K., Skjelbred, C. F., and Russell, M. B. (2011). Charcot-Marie-Tooth caused by a copy number variation in myelin protein zero. Eur. J. Med. Genet. 54, e580–e583. doi: 10.1016/j.ejmg.2011.06.006
Iapadre, G., Morana, G., Vari, M. S., Pinto, F., Lanteri, P., Tessa, A., et al. (2018). A novel homozygous MFN2 mutation associated with severe and atypical CMT2 phenotype. Eur. J. Paediatr. Neurol. 22, 563–567. doi: 10.1016/j.ejpn.2017.12.020
James, P. A., Cader, M. Z., Muntoni, F., Childs, A. M., Crow, Y. J., and Talbot, K. (2006). Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology 67, 1710–1712. doi: 10.1212/01.wnl.0000242619.52335.bc
Jin, S., Wang, W., Wang, R., Lv, H., Zhang, W., Wang, Z., et al. (2015). INF2 mutations associated with dominant inherited intermediate Charcot-Marie-Tooth neuropathy with focal segmental glomerulosclerosis in two Chinese patients. Clin. Neuropathol. 34, 275–281. doi: 10.5414/NP300835
Jordanova, A., Irobi, J., Thomas, F. P., Van Dijck, P., Meerschaert, K., Dewil, M., et al. (2006). Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 38, 197–202. doi: 10.1038/ng1727
Juneja, M., Burns, J., Saporta, M. A., and Timmerman, V. (2019). Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 90, 58–67. doi: 10.1136/jnnp-2018-318834
Kabzińska, D., Saifi, G. M., Drac, H., Rowińska-Marcińska, K., Hausmanowa-Petrusewicz, I., Kochański, A., et al. (2007). Charcot-Marie-Tooth disease type 4C4 caused by a novel Pro153Leu substitution in the GDAP1 gene. Acta Myol. 26, 108–111.
Kanwal, S., and Perveen, S. (2019). Association of SNP in JPH1 gene with severity of disease in Charcot Marie Tooth 2K patients. J. Pak. Med. Assoc. 69, 241–243.
Kijima, K., Numakura, C., Izumino, H., Umetsu, K., Nezu, A., Shiiki, T., et al. (2005). Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum. Genet. 116, 23–27. doi: 10.1007/s00439-004-1199-2
Kim, H. J., Hong, Y. B., Park, J. M., Choi, Y. R., Kim, Y. J., Yoon, B. R., et al. (2013). Mutations in the PLEKHG5 gene is relevant with autosomal recessive intermediate Charcot-Marie-Tooth disease. Orphanet. J. Rare. Dis. 8:104. doi: 10.1186/1750-1172-8-104
Kim, S. J., Nam, S. H., Kanwal, S., Nam, D. E., Yoo, D. H., Chae, J. H., et al. (2018). BAG3 mutation in a patient with atypical phenotypes of myofibrillar myopathy and Charcot-Marie-Tooth disease. Genes Genomics 40, 1269–1277. doi: 10.1007/s13258-018-0721-1
Kleffner, I., Schirmacher, A., Gess, B., Boentert, M., and Young, P. (2010). Four novel mutations of the myelin protein zero gene presenting as a mild and late-onset polyneuropathy. J. Neurol. 257, 1864–1868. doi: 10.1007/s00415-010-5624-2
Klein, C. J., Cunningham, J. M., Atkinson, E. J., Schaid, D. J., Hebbring, S. J., Anderson, S. A., et al. (2003). The gene for HMSN2C maps to 12q23-24: a region of neuromuscular disorders. Neurology 60, 1151–1156. doi: 10.1212/01.wnl.0000055900.30217.ea
Kochański, A., Kabzińska, D., Drac, H., Ryniewicz, B., Rowińska-Marcińska, K., and Hausmanowa-Petrusewicz, I. (2004). Early onset Charcot-Marie-Tooth type 1B disease caused by a novel Leu190fs mutation in the myelin protein zero gene. Eur. J. Paediatr. Neurol. 8, 221–224. doi: 10.1016/j.ejpn.2004.04.001
Korobova, F., Gauvin, T. J., and Higgs, H. N. (2014). A role for myosin II in mammalian mitochondrial fission. Curr. Biol. 24, 409–414. doi: 10.1016/j.cub.2013.12.032
Korobova, F., Ramabhadran, V., and Higgs, H. N. (2013). An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464–467. doi: 10.1126/science.1228360
Kotruchow, K., Kabzińska, D., Hausmanowa-Petrusewicz, I., and Kochański, A. (2013). A late-onset and mild form of Charcot-Marie-Tooth disease type 2 caused by a novel splice-site mutation within the Mitofusin-2 gene. Acta Myol. 32, 166–169.
Kurihara, S., Adachi, Y., Imai, C., Araki, H., Hattori, N., Numakura, C., et al. (2004). Charcot-Marie-Tooth families in Japan with MPZ Thr124Met mutation. J. Neurol. Neurosurg. Psychiatry 75, 1492–1494. doi: 10.1136/jnnp.2003.020107
Lagueny, A., Latour, P., Vital, A., Rajabally, Y., Le Masson, G., Ferrer, X., et al. (1999). Peripheral myelin modification in CMT1B correlates with MPZ gene mutations. Neuromuscul. Disord. 9, 361–367. doi: 10.1016/s0960-8966(99)00031-0
Larrea, D., Pera, M., Gonnelli, A., Quintana-Cabrera, R., Akman, H. O., Guardia-Laguarta, C., et al. (2019). MFN2 mutations in Charcot-Marie-Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum. Mol. Genet. 28, 1782–1800. doi: 10.1093/hmg/ddz008
Laššuthová, P., Brožková, D., Krůtová, M., Neupauerová, J., Haberlová, J., Mazanec, R., et al. (2015). Mutations in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics 16, 43–54. doi: 10.1007/s10048-014-0427-8
Lassuthova, P., Rebelo, A. P., Ravenscroft, G., Lamont, P. J., Davis, M. R., Manganelli, F., et al. (2018). Mutations in ATP1A1 cause dominant Charcot-Marie-Tooth Type 2. Am. J. Hum. Genet. 102, 505–514. doi: 10.1016/j.ajhg.2018.01.023
Latour, P., Blanquet, F., Nelis, E., Bonnebouche, C., Chapon, F., Diraison, P., et al. (1995). Mutations in the myelin protein zero gene associated with Charcot-Marie-Tooth disease type 1B. Hum. Mutat. 6, 50–54. doi: 10.1002/humu.1380060110
Laurà, M., Milani, M., Morbin, M., Moggio, M., Ripolone, M., Jann, S., et al. (2007). Rapid progression of late onset axonal Charcot-Marie-Tooth disease associated with a novel MPZ mutation in the extracellular domain. J. Neurol. Neurosurg. Psychiatry 78, 1263–1266. doi: 10.1136/jnnp.2006.112276
Lawson, V. H., Graham, B. V., and Flanigan, K. M. (2005). Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology 65, 197–204. doi: 10.1212/01.wnl.0000168898.76071.70
Leal, A., Berghoff, C., Berghoff, M., Rojas-Araya, M., Carolina, O., Heuss, D., et al. (2014). A costa rican family affected with Charcot-Marie-Tooth disease due to the myelin protein zero (MPZ) p.Thr124Met mutation shares the Belgian haplotype. Rev. Biol. Trop. 62, 1285–1293. doi: 10.15517/rbt.v62i4.13473
Leal, A., Bogantes-Ledezma, S., Ekici, A. B., Uebe, S., Thiel, C. T., Sticht, H., et al. (2018). The polynucleotide kinase 3’-phosphatase gene (PNKP) is involved in Charcot-Marie-Tooth disease (CMT2B2) previously related to MED25. Neurogenetics 19, 215–225. doi: 10.1007/s10048-018-0555-7
Leal, A., Huehne, K., Bauer, F., Sticht, H., Berger, P., Suter, U., et al. (2009). Identification of the variant Ala335Val of MED25 as responsible for CMT2B2: molecular data, functional studies of the SH3 recognition motif and correlation between wild-type MED25 and PMP22 RNA levels in CMT1A animal models. Neurogenetics 10, 275–287. doi: 10.1007/s10048-009-0183-3
Leal, N. S., Schreiner, B., Pinho, C. M., Filadi, R., Wiehager, B., Karlstrom, H., et al. (2016). Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J. Cell Mol. Med. 20, 1686–1695. doi: 10.1111/jcmm.12863
Lee, M., Park, C. H., Chung, H. K., Kim, H. J., Choi, Y., Yoo, J. H., et al. (2017). Cerebral white matter abnormalities in patients with charcot-marie-tooth disease. Ann. Neurol. 81, 147–151. doi: 10.1002/ana.24824
Lee, Y. C., Yu, C. T., Lin, K. P., Chang, M. H., Hsu, S. L., Liu, Y. F., et al. (2008). MPZ mutation G123S characterization: evidence for a complex pathogenesis in CMT disease. Neurology 70, 273–277. doi: 10.1212/01.wnl.0000296828.66915.bf
Liang, J. J., Grogan, M., Ackerman, M. J., and Goodsell, K. (2016). LMNA-mediated arrhythmogenic right ventricular cardiomyopathy and Charcot-Marie-Tooth Type 2B1: a patient-discovered unifying diagnosis. J. Cardiovasc. Electrophysiol. 27, 868–871. doi: 10.1111/jce.12984
Liu, X. M., Tang, B. S., Zhao, G. H., Xia, K., Zhang, F. F., Pan, Q., et al. (2005). [Mutation analysis of small heat shock protein 27 gene in Chinese patients with Charcot-Marie-Tooth disease]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 22, 510–513.
Loiseau, D., Chevrollier, A., Verny, C., Guillet, V., Gueguen, N., Pou de Crescenzo, M. A., et al. (2007). Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann. Neurol. 61, 315–323. doi: 10.1002/ana.21086
Luo, W., Tang, B., Zhao, G., Li, Q., Xiao, J., Yang, Q., et al. (2003). [Mutation analysis of neurofilament-light gene in Chinese Charcot-Marie-Tooth disease]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 20, 169–170.
Lupo, V., García-García, F., Sancho, P., Tello, C., García-Romero, M., Villarreal, L., et al. (2016). Assessment of targeted next-generation sequencing as a tool for the diagnosis of Charcot-Marie-Tooth disease and hereditary motor neuropathy. J. Mol. Diagn. 18, 225–234. doi: 10.1016/j.jmoldx.2015.10.005
Lupski, J. R. (2000). Axonal Charcot-Marie-Tooth disease and the neurofilament light gene (NF-L). Am. J. Hum. Genet. 67, 8–10. doi: 10.1086/302986
Lv, H., Wang, L., Li, W., Qiao, X., Li, Y., Wang, Z., et al. (2013). Mitofusin 2 gene mutation causing early-onset CMT2A with different progressive courses. Clin. Neuropathol. 32, 16–23. doi: 10.5414/np300464
Lv, H., Wang, L., Zhang, W., Wang, Z., Zuo, Y., Liu, J., et al. (2015). A cohort study of Han Chinese MFN2-related Charcot-Marie-Tooth 2A. J. Neurol. Sci. 358, 153–157. doi: 10.1016/j.jns.2015.08.1528
Mademan, I., Deconinck, T., Dinopoulos, A., Voit, T., Schara, U., Devriendt, K., et al. (2013). De novo INF2 mutations expand the genetic spectrum of hereditary neuropathy with glomerulopathy. Neurology 81, 1953–1958. doi: 10.1212/01.wnl.0000436615.58705.c9
Maeda, M. H., Mitsui, J., Soong, B. W., Takahashi, Y., Ishiura, H., Hayashi, S., et al. (2012). Increased gene dosage of myelin protein zero causes Charcot-Marie-Tooth disease. Ann. Neurol. 71, 84–92. doi: 10.1002/ana.22658
Mai, P. T., Le, D. T., Nguyen, T. T., Le Gia, H. L., Nguyen Le, T. H., Le, M., et al. (2019). Novel GDAP1 mutation in a vietnamese family with Charcot-Marie-Tooth Disease. Biomed. Res. Int. 2019:7132494. doi: 10.1155/2019/7132494
Manganelli, F., Nolano, M., Pisciotta, C., Provitera, V., Fabrizi, G. M., Cavallaro, T., et al. (2015). Charcot-Marie-Tooth disease: new insights from skin biopsy. Neurology 85, 1202–1208. doi: 10.1212/wnl.0000000000001993
Manor, U., Bartholomew, S., Golani, G., Christenson, E., Kozlov, M., Higgs, H., et al. (2015). A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 4:e08828. doi: 10.7554/eLife.08828
Martí, S., León, M., Orellana, C., Prieto, J., Ponsoda, X., López-García, C., et al. (2017). Generation of a disease-specific iPS cell line derived from a patient with Charcot-Marie-Tooth type 2K lacking functional GDAP1 gene. Stem Cell Res. 18, 1–4. doi: 10.1016/j.scr.2016.11.017
Martin, A. M., Maradei, S. J., and Velasco, H. M. (2015). Charcot Marie Tooth disease (CMT4A) due to GDAP1 mutation: report of a Colombian family. Colomb. Med. 46, 194–198.
Marttila, M., Rautenstrauss, B., Huehne, K., Laitinen, V., Majamaa, K., and Kärppä, M. (2012). A novel mutation of myelin protein zero associated with late-onset predominantly axonal Charcot-Marie-Tooth disease. J. Neurol. 259, 1585–1589. doi: 10.1007/s00415-011-6382-5
Masingue, M., Perrot, J., Carlier, R. Y., Piguet-Lacroix, G., Latour, P., and Stojkovic, T. (2018). WES homozygosity mapping in a recessive form of Charcot-Marie-Tooth neuropathy reveals intronic GDAP1 variant leading to a premature stop codon. Neurogenetics 19, 67–76. doi: 10.1007/s10048-018-0539-7
Matsumoto, N., Hori, I., Murase, T., Tsuji, T., Miyake, S., Inatani, M., et al. (2020). Intermitochondrial signaling regulates the uniform distribution of stationary mitochondria in axons. bioRxiv [Preprint]. doi: 10.1101/2020.07.31.230250 [Preprint],
Mazzeo, A., Muglia, M., Rodolico, C., Toscano, A., Patitucci, A., Quattrone, A., et al. (2008). Charcot-Marie-Tooth disease type 1B: marked phenotypic variation of the Ser78Leu mutation in five Italian families. Acta Neurol. Scand. 118, 328–332. doi: 10.1111/j.1600-0404.2008.01021.x
McCorquodale, D. S. III, Montenegro, G., Peguero, A., Carlson, N., Speziani, F., Price, J., et al. (2011). Mutation screening of mitofusin 2 in Charcot-Marie-Tooth disease type 2. J. Neurol. 258, 1234–1239. doi: 10.1007/s00415-011-5910-7
McLaughlin, H. M., Sakaguchi, R., Giblin, W., Wilson, T. E., Biesecker, L., Lupski, J. R., et al. (2012). A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum. Mutat. 33, 244–253. doi: 10.1002/humu.21635
McLaughlin, H. M., Sakaguchi, R., Liu, C., Igarashi, T., Pehlivan, D., Chu, K., et al. (2010). Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet. 87, 560–566. doi: 10.1016/j.ajhg.2010.09.008
McLelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 7:e32866. doi: 10.7554/eLife.32866
Meggouh, F., Bienfait, H. M., Weterman, M. A., de Visser, M., and Baas, F. (2006). Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology 67, 1476–1478. doi: 10.1212/01.wnl.0000240068.21499.f5
Milley, G. M., Varga, E. T., Grosz, Z., Nemes, C., Arányi, Z., Boczán, J., et al. (2018). Genotypic and phenotypic spectrum of the most common causative genes of Charcot-Marie-Tooth disease in Hungarian patients. Neuromuscul. Disord. 28, 38–43. doi: 10.1016/j.nmd.2017.08.007
Miltenberger-Miltenyi, G., Janecke, A. R., Wanschitz, J. V., Timmerman, V., Windpassinger, C., Auer-Grumbach, M., et al. (2007). Clinical and electrophysiological features in Charcot-Marie-Tooth disease with mutations in the NEFL gene. Arch. Neurol. 64, 966–970. doi: 10.1001/archneur.64.7.966
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666. doi: 10.1016/j.neuron.2017.09.055
Miura, S., Morikawa, T., Fujioka, R., Noda, K., Kosaka, K., Taniwaki, T., et al. (2017). A novel missense variant (Gln220Arg) of GNB4 encoding guanine nucleotide-binding protein, subunit beta-4 in a Japanese family with autosomal dominant motor and sensory neuropathy. Eur. J. Med. Genet. 60, 474–478. doi: 10.1016/j.ejmg.2017.06.006
Montecchiani, C., Pedace, L., Lo Giudice, T., Casella, A., Mearini, M., Gaudiello, F., et al. (2016). ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain 139(Pt 1), 73–85. doi: 10.1093/brain/awv320
Morelli, K. H., Seburn, K. L., Schroeder, D. G., Spaulding, E. L., Dionne, L. A., Cox, G. A., et al. (2017). Severity of demyelinating and axonal neuropathy mouse models is modified by genes affecting structure and function of peripheral nodes. Cell Rep. 18, 3178–3191. doi: 10.1016/j.celrep.2017.03.009
Moroni, I., Morbin, M., Milani, M., Ciano, C., Bugiani, M., Pagliano, E., et al. (2009). Novel mutations in the GDAP1 gene in patients affected with early-onset axonal Charcot-Marie-Tooth type 4A. Neuromuscul. Disord. 19, 476–480. doi: 10.1016/j.nmd.2009.04.014
Motley, W. W., Seburn, K. L., Nawaz, M. H., Miers, K. E., Cheng, J., Antonellis, A., et al. (2011). Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet. 7:e1002399. doi: 10.1371/journal.pgen.1002399
Muglia, M., Vazza, G., Patitucci, A., Milani, M., Pareyson, D., Taroni, F., et al. (2007). A novel founder mutation in the MFN2 gene associated with variable Charcot-Marie-Tooth type 2 phenotype in two families from Southern Italy. J. Neurol. Neurosurg. Psychiatry 78, 1286–1287. doi: 10.1136/jnnp.2007.115774
Nakagawa, M., and Takashima, H. (2003). [Molecular mechanisms of hereditary neuropathy: genotype-phenotype correlation]. Rinsho Byori 51, 536–543.
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U.S.A. 113, 11249–11254. doi: 10.1073/pnas.1606786113
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2017). Reply to Filadi et al.: does Mitofusin 2 tether or separate endoplasmic reticulum and mitochondria? Proc. Natl. Acad. Sci. U.S.A. 114, E2268–E2269. doi: 10.1073/pnas.1618610114
Nelis, E., Timmerman, V., De Jonghe, P., Vandenberghe, A., Pham-Dinh, D., Dautigny, A., et al. (1994). Rapid screening of myelin genes in CMT1 patients by SSCP analysis: identification of new mutations and polymorphisms in the P0 gene. Hum. Genet. 94, 653–657. doi: 10.1007/bf00206959
Neupauerová, J., Grečmalová, D., Seeman, P., and Laššuthová, P. (2016). Massively Parallel Sequencing Detected a Mutation in the MFN2 Gene Missed by Sanger Sequencing Due to a Primer Mismatch on an SNP Site. Ann. Hum. Genet. 80, 182–186. doi: 10.1111/ahg.12151
Neusch, C., Senderek, J., Eggermann, T., Elolff, E., Bahr, M., and Schneider-Gold, C. (2007). Mitofusin 2 gene mutation (R94Q) causing severe early-onset axonal polyneuropathy (CMT2A). Eur. J. Neurol. 14, 575–577. doi: 10.1111/j.1468-1331.2006.01688.x
Newman, L. E., and Shadel, G. S. (2018). Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J. Cell Biol. 217, 3327–3329. doi: 10.1083/jcb.201808118
Nicholson, G., Kennerson, M., Brewer, M., Garbern, J., and Shy, M. (2009). Genotypes & sensory phenotypes in 2 new X-linked neuropathies (CMTX3 and dSMAX) and dominant CMT/HMN overlap syndromes. Adv. Exp. Med. Biol. 652, 201–206. doi: 10.1007/978-90-481-2813-6_13
Nicolaou, P., Cianchetti, C., Minaidou, A., Marrosu, G., Zamba-Papanicolaou, E., Middleton, L., et al. (2013). A novel LRSAM1 mutation is associated with autosomal dominant axonal Charcot-Marie-Tooth disease. Eur. J. Hum. Genet. 21, 190–194. doi: 10.1038/ejhg.2012.146
Ouvrier, R., and Grew, S. (2010). Mechanisms of disease and clinical features of mutations of the gene for mitofusin 2: an important cause of hereditary peripheral neuropathy with striking clinical variability in children and adults. Dev. Med. Child. Neurol. 52, 328–330. doi: 10.1111/j.1469-8749.2010.03613.x
Pan, Z. N., Pan, M. H., Sun, M. H., Li, X. H., Zhang, Y., and Sun, S. C. (2020). RAB7 GTPase regulates actin dynamics for DRP1-mediated mitochondria function and spindle migration in mouse oocyte meiosis. FASEB J. 34, 9615–9627. doi: 10.1096/fj.201903013R
Pareyson, D., and Marchesi, C. (2009). Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 8, 654–667. doi: 10.1016/s1474-4422(09)70110-3
Park, H. J., Kim, H. J., Hong, Y. B., Nam, S. H., Chung, K. W., and Choi, B. O. (2014). A novel INF2 mutation in a Korean family with autosomal dominant intermediate Charcot-Marie-Tooth disease and focal segmental glomerulosclerosis. J. Peripher. Nerv. Syst. 19, 175–179. doi: 10.1111/jns5.12062
Park, S. Y., Kim, S. Y., Hong, Y. H., Cho, S. I., Seong, M. W., and Park, S. S. (2012). A novel double mutation in cis in MFN2 causes Charcot-Marie-Tooth neuropathy type 2A. Neurogenetics 13, 275–280. doi: 10.1007/s10048-012-0327-8
Pathak, D., Sepp, K. J., and Hollenbeck, P. J. (2010). Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci. 30, 8984–8992. doi: 10.1523/jneurosci.1621-10.2010
Pehlivan, D., Coban Akdemir, Z., Karaca, E., Bayram, Y., Jhangiani, S., Yildiz, E. P., et al. (2015). Exome sequencing reveals homozygous TRIM2 mutation in a patient with early onset CMT and bilateral vocal cord paralysis. Hum. Genet. 134, 671–673. doi: 10.1007/s00439-015-1548-3
Pham-Dinh, D., Fourbil, Y., Blanquet, F., Mattéi, M. G., Roeckel, N., Latour, P., et al. (1993). The major peripheral myelin protein zero gene: structure and localization in the cluster of Fc gamma receptor genes on human chromosome 1q21.3-q23. Hum. Mol. Genet. 2, 2051–2054. doi: 10.1093/hmg/2.12.2051
Pitceathly, R. D., Murphy, S. M., Cottenie, E., Chalasani, A., Sweeney, M. G., Woodward, C., et al. (2012). Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology 79, 1145–1154. doi: 10.1212/WNL.0b013e3182698d8d
Pla-Martín, D., Calpena, E., Lupo, V., Márquez, C., Rivas, E., Sivera, R., et al. (2015). Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 24, 213–229. doi: 10.1093/hmg/ddu440
Qin, L., Yang, C., Lü, T., Li, L., Zong, D., and Wu, Y. (2019). [Analysis of GDAP1 gene mutation in a pedigree with autosomal dominant Charcot-Marie-Tooth disease]. Nan Fang Yi Ke Da Xue Xue Bao 39, 63–68. doi: 10.12122/j.issn.1673-4254.2019.01.10
Quattrini, A., Feltri, M. L., Previtali, S., Fasolini, M., Messing, A., and Wrabetz, L. (1999). Peripheral nerve dysmyelination due to P0 glycoprotein overexpression is dose-dependent. Ann. N. Y. Acad. Sci. 883, 294–301.
Quintero, O. A., DiVito, M. M., Adikes, R. C., Kortan, M. B., Case, L. B., Lier, A. J., et al. (2009). Human Myo19 is a novel myosin that associates with mitochondria. Curr. Biol. 19, 2008–2013. doi: 10.1016/j.cub.2009.10.026
Rangaraju, V., Lauterbach, M., and Schuman, E. M. (2019). Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell 176, 73.e15–84.e15. doi: 10.1016/j.cell.2018.12.013
Roa, B. B., Warner, L. E., Garcia, C. A., Russo, D., Lovelace, R., Chance, P. F., et al. (1996). Myelin protein zero (MPZ) gene mutations in nonduplication type 1 Charcot-Marie-Tooth disease. Hum. Mutat. 7, 36–45. doi: 10.1002/(sici)1098-100419967:1<36::Aid-humu5<3.0.Co;2-n
Rocha, A. G., Franco, A., Krezel, A. M., Rumsey, J. M., Alberti, J. M., Knight, W. C., et al. (2018). MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 360, 336–341. doi: 10.1126/science.aao1785
Rodriguez, P. Q., Lohkamp, B., Celsi, G., Mache, C. J., Auer-Grumbach, M., Wernerson, A., et al. (2013). Novel INF2 mutation p. L77P in a family with glomerulopathy and Charcot-Marie-Tooth neuropathy. Pediatr. Nephrol. 28, 339–343. doi: 10.1007/s00467-012-2299-1
Rosberg, M. R., Alvarez, S., Krarup, C., and Moldovan, M. (2013). Functional recovery of regenerating motor axons is delayed in mice heterozygously deficient for the myelin protein P(0) gene. Neurochem. Res. 38, 1266–1277. doi: 10.1007/s11064-013-1030-3
Rougeot, C., Chabrier, S., Camdessanche, J. P., Prieur, F., d’Anjou, M. C., and Latour, P. (2008). Clinical, electrophysiological and genetic studies of two families with mutations in the GDAP1 gene. Neuropediatrics 39, 184–187. doi: 10.1055/s-0028-1085467
Rudnik-Schöneborn, S., Tölle, D., Senderek, J., Eggermann, K., Elbracht, M., Kornak, U., et al. (2016). Diagnostic algorithms in Charcot-Marie-Tooth neuropathies: experiences from a German genetic laboratory on the basis of 1206 index patients. Clin. Genet. 89, 34–43. doi: 10.1111/cge.12594
Rzepnikowska, W., and Kochański, A. (2018). A role for the GDAP1 gene in the molecular pathogenesis of Charcot-Marie-Tooth disease. Acta Neurobiol. Exp. 78, 1–13. doi: 10.21307/ane-2018-002
Sabet, A., Li, J., Ghandour, K., Pu, Q., Wu, X., Kamholz, J., et al. (2006). Skin biopsies demonstrate MPZ splicing abnormalities in Charcot-Marie-Tooth neuropathy 1B. Neurology 67, 1141–1146. doi: 10.1212/01.wnl.0000238499.37764.b1
Saghira, C., Bis, D. M., Stanek, D., Strickland, A., Herrmann, D. N., Reilly, M. M., et al. (2018). Variant pathogenicity evaluation in the community-driven Inherited Neuropathy Variant Browser. Hum. Mutat. 39, 635–642. doi: 10.1002/humu.23412
Sahin-Calapoglu, N., Soyoz, M., Calapoglu, M., and Ozcelik, N. (2009a). Genetic study of demyelinating form of autosomal-recessive Charcot-Marie-tooth diseases in a Turkish family. Int. J. Neurosci. 119, 1179–1189. doi: 10.1080/00207450902869906
Sahin-Calapoglu, N., Tan, M., Soyoz, M., Calapoglu, M., and Ozcelik, N. (2009b). Novel GDAP1 mutation in a Turkish family with CMT2K (CMT2K with novel GDAP1 mutation). Neuromol. Med. 11, 106–113. doi: 10.1007/s12017-009-8062-5
Sanmaneechai, O., Feely, S., Scherer, S. S., Herrmann, D. N., Burns, J., Muntoni, F., et al. (2015). Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain 138(Pt 11), 3180–3192. doi: 10.1093/brain/awv241
Saporta, M. A., Dang, V., Volfson, D., Zou, B., Xie, X. S., Adebola, A., et al. (2015). Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 263, 190–199. doi: 10.1016/j.expneurol.2014.10.005
Schiavon, C. R., Zhang, T., Zhao, B., Moore, A. S., Wales, P., Andrade, L. R., et al. (2020). Actin chromobody imaging reveals sub-organellar actin dynamics. Nat. Methods 17, 917–921. doi: 10.1038/s41592-020-0926-5
Senderek, J., Bergmann, C., Ramaekers, V. T., Nelis, E., Bernert, G., Makowski, A., et al. (2003). Mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot-Marie-Tooth neuropathy. Brain 126(Pt 3), 642–649. doi: 10.1093/brain/awg068
Senderek, J., Hermanns, B., Lehmann, U., Bergmann, C., Marx, G., Kabus, C., et al. (2000). Charcot-Marie-Tooth neuropathy type 2 and P0 point mutations: two novel amino acid substitutions (Asp61Gly; Tyr119Cys) and a possible “hotspot”” on Thr124Met. Brain Pathol. 10, 235–248. doi: 10.1111/j.1750-3639.2000.tb00257.x
Shin, J. S., Chung, K. W., Cho, S. Y., Yun, J., Hwang, S. J., Kang, S. H., et al. (2008). NEFL Pro22Arg mutation in Charcot-Marie-Tooth disease type 1. J. Hum. Genet. 53, 936–940. doi: 10.1007/s10038-008-0333-8
Shy, M., Rebelo, A. P., Feely, S. M., Abreu, L. A., Tao, F., Swenson, A., et al. (2018). Mutations in BAG3 cause adult-onset Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 89, 313–315. doi: 10.1136/jnnp-2017-315929
Silander, K., Meretoja, P., Nelis, E., Timmerman, V., Van Broeckhoven, C., Aula, P., et al. (1996). A de novo duplication in 17p11.2 and a novel mutation in the Po gene in two Déjérine-Sottas syndrome patients. Hum. Mutat. 8, 304–310. doi: 10.1002/(sici)1098-100419968:4<304::Aid-humu2<3.0.Co;2-7
Skre, H. (1974). Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 6, 98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x
Solla, P., Vannelli, A., Bolino, A., Marrosu, G., Coviello, S., Murru, M. R., et al. (2010). Heat shock protein 27 R127W mutation: evidence of a continuum between axonal Charcot-Marie-Tooth and distal hereditary motor neuropathy. J. Neurol. Neurosurg. Psychiatry 81, 958–962. doi: 10.1136/jnnp.2009.181636
Soong, B. W., Huang, Y. H., Tsai, P. C., Huang, C. C., Pan, H. C., Lu, Y. C., et al. (2013). Exome sequencing identifies GNB4 mutations as a cause of dominant intermediate Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 92, 422–430. doi: 10.1016/j.ajhg.2013.01.014
Speevak, M. D., and Farrell, S. A. (2013). Charcot-Marie-Tooth 1B caused by expansion of a familial myelin protein zero (MPZ) gene duplication. Eur. J. Med. Genet. 56, 566–569. doi: 10.1016/j.ejmg.2013.06.004
Sugiura, A., Nagashima, S., Tokuyama, T., Amo, T., Matsuki, Y., Ishido, S., et al. (2013). MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell 51, 20–34. doi: 10.1016/j.molcel.2013.04.023
Sun, S. C., Ma, D., Li, M. Y., Zhang, R. X., Huang, C., Huang, H. J., et al. (2019). Mutations in C1orf194, encoding a calcium regulator, cause dominant Charcot-Marie-Tooth disease. Brain 142, 2215–2229. doi: 10.1093/brain/awz151
Sun, Y., Hu, G., Luo, J., Fang, D., Yu, Y., Wang, X., et al. (2017). Mutations in methionyl-tRNA synthetase gene in a Chinese family with interstitial lung and liver disease, postnatal growth failure and anemia. J. Hum. Genet. 62, 647–651. doi: 10.1038/jhg.2017.10
Tamiya, G., Makino, S., Hayashi, M., Abe, A., Numakura, C., Ueki, M., et al. (2014). A mutation of COX6A1 causes a recessive axonal or mixed form of Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 95, 294–300. doi: 10.1016/j.ajhg.2014.07.013
Tan, C. A., Rabideau, M., Blevins, A., Westbrook, M. J., Ekstein, T., Nykamp, K., et al. (2016). Autosomal recessive MFN2-related Charcot-Marie-Tooth disease with diaphragmatic weakness: case report and literature review. Am. J. Med. Genet. A 170, 1580–1584. doi: 10.1002/ajmg.a.37611
Tang, B., Liu, X., Zhao, G., Luo, W., Xia, K., Pan, Q., et al. (2005). Mutation analysis of the small heat shock protein 27 gene in chinese patients with Charcot-Marie-Tooth disease. Arch. Neurol. 62, 1201–1207. doi: 10.1001/archneur.62.8.1201
Tazir, M., Bellatache, M., Nouioua, S., and Vallat, J. M. (2013). Autosomal recessive Charcot-Marie-Tooth disease: from genes to phenotypes. J. Peripher. Nerv. Syst. 18, 113–129. doi: 10.1111/jns5.12026
Tétreault, M., Gonzalez, M., Dicaire, M. J., Allard, P., Gehring, K., Leblanc, D., et al. (2015). Adult-onset painful axonal polyneuropathy caused by a dominant NAGLU mutation. Brain 138(Pt 6), 1477–1483. doi: 10.1093/brain/awv074
Tokuda, N., Noto, Y., Kitani-Morii, F., Hamano, A., Kasai, T., Shiga, K., et al. (2015). Parasympathetic dominant autonomic dysfunction in Charcot-Marie-Tooth Disease Type 2J with the MPZ Thr124Met Mutation. Intern. Med. 54, 1919–1922. doi: 10.2169/internalmedicine.54.4259
Trevisan, T., Pendin, D., Montagna, A., Bova, S., Ghelli, A. M., and Daga, A. (2018). Manipulation of mitochondria dynamics reveals separate roles for form and function in mitochondria distribution. Cell Rep. 23, 1742–1753. doi: 10.1016/j.celrep.2018.04.017
Vallat, J. M., Mathis, S., and Funalot, B. (2013). The various Charcot-Marie-Tooth diseases. Curr. Opin. Neurol. 26, 473–480. doi: 10.1097/WCO.0b013e328364c04b
Vallat, J. M., Ouvrier, R. A., Pollard, J. D., Magdelaine, C., Zhu, D., Nicholson, G. A., et al. (2008). Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with mitofusin 2 mutations. J. Neuropathol. Exp. Neurol. 67, 1097–1102. doi: 10.1097/NEN.0b013e31818b6cbc
Venkatesh, K., Mathew, A., and Koushika, S. P. (2019). Role of actin in organelle trafficking in neurons. Cytoskeleton 77, 97–109. doi: 10.1002/cm.21580
Verhoeven, K., Claeys, K. G., Zuchner, S., Schroder, J. M., Weis, J., Ceuterick, C., et al. (2006). MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain 129(Pt 8), 2093–2102. doi: 10.1093/brain/awl126
Verhoeven, K., De Jonghe, P., Coen, K., Verpoorten, N., Auer-Grumbach, M., Kwon, J. M., et al. (2003). Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 72, 722–727. doi: 10.1086/367847
Vielhaber, S., Debska-Vielhaber, G., Peeva, V., Schoeler, S., Kudin, A. P., Minin, I., et al. (2013). Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 125, 245–256. doi: 10.1007/s00401-012-1036-y
Vital, A., Latour, P., Sole, G., Ferrer, X., Rouanet, M., Tison, F., et al. (2012). A French family with Charcot-Marie-Tooth disease related to simultaneous heterozygous MFN2 and GDAP1 mutations. Neuromuscul. Disord. 22, 735–741. doi: 10.1016/j.nmd.2012.04.001
Wagner, J. D., Huang, L., Tetreault, M., Majewski, J., Boycott, K. M., Bulman, D. E., et al. (2015). Autosomal recessive axonal polyneuropathy in a sibling pair due to a novel homozygous mutation in IGHMBP2. Neuromuscul. Disord. 25, 794–799. doi: 10.1016/j.nmd.2015.07.017
Wang, R., He, J., Li, J. J., Ni, W., Wu, Z. Y., Chen, W. J., et al. (2015). Clinical and genetic spectra in a series of Chinese patients with Charcot-Marie-Tooth disease. Clin. Chim. Acta 451(Pt B), 263–270. doi: 10.1016/j.cca.2015.10.007
Weedon, M. N., Hastings, R., Caswell, R., Xie, W., Paszkiewicz, K., Antoniadi, T., et al. (2011). Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 89, 308–312. doi: 10.1016/j.ajhg.2011.07.002
Weeks, S. D., Muranova, L. K., Heirbaut, M., Beelen, S., Strelkov, S. V., and Gusev, N. B. (2018). Characterization of human small heat shock protein HSPB1 α-crystallin domain localized mutants associated with hereditary motor neuron diseases. Sci. Rep. 8:688. doi: 10.1038/s41598-017-18874-x
Werheid, F., Azzedine, H., Zwerenz, E., Bozkurt, A., Moeller, M. J., Lin, L., et al. (2016). Underestimated associated features in CMT neuropathies: clinical indicators for the causative gene? Brain Behav. 6:e00451. doi: 10.1002/brb3.451
West, A. P. (2017). Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 391, 54–63. doi: 10.1016/j.tox.2017.07.016
Weterman, M. A. J., Kuo, M., Kenter, S. B., Gordillo, S., Karjosukarso, D. W., Takase, R., et al. (2018). Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities. Hum. Mol. Genet. 27, 4036–4050. doi: 10.1093/hmg/ddy290
Wong, Y. C., Peng, W., and Krainc, D. (2019). Lysosomal regulation of inter-mitochondrial contact fate and motility in Charcot-Marie-Tooth Type 2. Dev. Cell 50, 339.e4–354.e4. doi: 10.1016/j.devcel.2019.05.033
Wong, Y. C., Ysselstein, D., and Krainc, D. (2018). Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386. doi: 10.1038/nature25486
Xie, W., Nangle, L. A., Zhang, W., Schimmel, P., and Yang, X. L. (2007). Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 104, 9976–9981. doi: 10.1073/pnas.0703908104
Xie, W., Schimmel, P., and Yang, X. L. (2006). Crystallization and preliminary X-ray analysis of a native human tRNA synthetase whose allelic variants are associated with Charcot-Marie-Tooth disease. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62(Pt 12), 1243–1246. doi: 10.1107/s1744309106046434
Xie, Y., Li, X., Liu, L., Hu, Z., Huang, S., Zhan, Y., et al. (2016). MFN2-related genetic and clinical features in a cohort of Chinese CMT2 patients. J. Peripher. Nerv. Syst. 21, 38–44. doi: 10.1111/jns.12159
Xin, B., Puffenberger, E., Nye, L., Wiznitzer, M., and Wang, H. (2008). A novel mutation in the GDAP1 gene is associated with autosomal recessive Charcot-Marie-Tooth disease in an Amish family. Clin. Genet. 74, 274–278. doi: 10.1111/j.1399-0004.2008.01018.x
Xu, J. L., Zhang, Y., Zhao, C. Y., Jiang, P. F., Yuan, Z. F., Yu, Y. L., et al. (2019). [A genotyping study of 13 cases of early-onset Charcot-Marie-Tooth disease]. Zhongguo Dang Dai Er Ke Za Zhi 21, 670–675. doi: 10.7499/j.issn.1008-8830.2019.07.010
Yamaguchi, M., and Takashima, H. (2018). Drosophila charcot-marie-tooth disease models. Adv. Exp. Med. Biol. 1076, 97–117. doi: 10.1007/978-981-13-0529-0_7
Yang, C., and Svitkina, T. M. (2019). Ultrastructure and dynamics of the actin-myosin II cytoskeleton during mitochondrial fission. Nat. Cell Biol. 21, 603–613. doi: 10.1038/s41556-019-0313-6
Yoshimura, A., Yuan, J. H., Hashiguchi, A., Hiramatsu, Y., Ando, M., Higuchi, Y., et al. (2017). Clinical and mutational spectrum of Japanese patients with Charcot-Marie-Tooth disease caused by GDAP1 variants. Clin. Genet. 92, 274–280. doi: 10.1111/cge.13002
Yu, S. B., and Pekkurnaz, G. (2018). Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J. Mol. Biol. 430, 3922–3941. doi: 10.1016/j.jmb.2018.07.027
Yuan, J. H., Hashiguchi, A., Yoshimura, A., Yaguchi, H., Tsuzaki, K., Ikeda, A., et al. (2017). Clinical diversity caused by novel IGHMBP2 variants. J. Hum. Genet. 62, 599–604. doi: 10.1038/jhg.2017.15
Zhang, R. X., Guo, P., Ren, Z. J., Zhao, G. H., Liu, S. M., Liu, T., et al. (2010). [Mutation analysis of LITAF, RAB7, LMNA and MTMR2 genes in Chinese Charcot-Marie-Tooth disease.]. Yi Chuan 32, 817–823. doi: 10.3724/sp.j.1005.2010.00817
Zhang, R. X., Tang, B. S., Zi, X. H., Luo, W., Xia, K., Pan, Q., et al. (2004). [Mutation analysis of ganglioside-induced differentiation associated protein-1 gene in Chinese Charcot-Marie-Tooth disease]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 21, 207–210.
Zhao, T., Huang, X., Han, L., Wang, X., Cheng, H., Zhao, Y., et al. (2012). Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J. Biol. Chem. 287, 23615–23625. doi: 10.1074/jbc.M112.379164
Zhao, Z., Hashiguchi, A., Hu, J., Sakiyama, Y., Okamoto, Y., Tokunaga, S., et al. (2012). Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology 78, 1644–1649. doi: 10.1212/WNL.0b013e3182574f8f
Zhao, Z. H., Chen, Z. T., Zhou, R. L., and Wang, Y. Z. (2019). A Chinese pedigree with a novel mutation in GJB1 gene and a rare variation in DHTKD1 gene for diverse Charcot-Marie-Tooth diseases. Mol. Med. Rep. 19, 4484–4490. doi: 10.3892/mmr.2019.10058
Zimoń, M., Baets, J., Fabrizi, G. M., Jaakkola, E., Kabzińska, D., Pilch, J., et al. (2011). Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77, 540–548. doi: 10.1212/WNL.0b013e318228fc70
Zimoń, M., Battaloğlu, E., Parman, Y., Erdem, S., Baets, J., De Vriendt, E., et al. (2015). Unraveling the genetic landscape of autosomal recessive Charcot-Marie-Tooth neuropathies using a homozygosity mapping approach. Neurogenetics 16, 33–42. doi: 10.1007/s10048-014-0422-0
Zuchner, S., De Jonghe, P., Jordanova, A., Claeys, K. G., Guergueltcheva, V., Cherninkova, S., et al. (2006). Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 59, 276–281. doi: 10.1002/ana.20797
Keywords: organelle transport, axonal transport deficiency, neurodegeneration, cytoskeleton, mitochondria, Charcot-Marie-Tooth (CMT) disease
Citation: Schiavon CR, Shadel GS and Manor U (2021) Impaired Mitochondrial Mobility in Charcot-Marie-Tooth Disease. Front. Cell Dev. Biol. 9:624823. doi: 10.3389/fcell.2021.624823
Received: 01 November 2020; Accepted: 05 January 2021;
Published: 01 February 2021.
Edited by:
Carlos M. Guardia, National Institutes of Health (NIH), United StatesReviewed by:
Masanori Nakagawa, Kyoto Prefectural University of Medicine, JapanRoman Polishchuk, Telethon Institute of Genetics and Medicine (TIGEM), Italy
Copyright © 2021 Schiavon, Shadel and Manor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uri Manor, umanor@salk.edu