Dr. William J. West first described infantile spasms in 1841 in a letter to The Lancet, in which he conveyed, “(the) case I have witnessed is in my own child.”1 He vividly depicts several key manifestations of the eponymous West syndrome, later defined by a triad of epileptic spasms (ES), developmental arrest or regression, and hypsarrhythmia on EEG seen in infants. ES are a distinctive seizure type characterized by sudden and brief tonic contractions (flexion, extension, or mixed) of axial musculature and extremities, lasting 1 to 2 seconds. Multiple ES commonly occur every few seconds within clusters lasting several minutes. Within a cluster, the apparent frequency and intensity of ES tend to follow a crescendo-decrescendo pattern. Clusters typically occur at least once per day and are most frequent upon awakening, although an ES can occur in an isolated fashion and at any time of day.
ES can occur at any age, although the infantile spasms of West syndrome are part of a particularly inauspicious, age-specific epileptic encephalopathy. The age at onset is 1-24 months, with a peak between 3-12 months, at a time when the infant typically begins to reach out, explore, and absorb information about the world. Development often plateaus at ES onset, and previously achieved milestones may be lost soon after. Prognosis often is poor, but if the disorder can be recognized promptly and treated quickly, normalization of development trajectory and cognitive potential is possible.2
Hypsarrhythmia is a pathognomonic interictal pattern seen on EEG with chaotic, high-amplitude, and multifocal epileptiform discharges. However, hypsarrhythmia may be intermittent, incomplete in all elements (eg, abundant multifocal discharges without a chaotic background), or absent, especially early in the course of disease. Likewise, developmental impairment may be subtle or not apparent early. Diagnostic uncertainty without the complete triad of findings in West syndrome may lead to delayed treatment, to the detriment of developmental outcomes.3 The ILAE has proposed a more inclusive diagnosis of infantile epileptic spasms syndrome (IESS) for any infant with ES and either developmental impairment or hypsarrhythmia.4
Differential Diagnosis
A typical clinical history and classic appearance of ES can facilitate early IESS diagnosis in many cases. However, ES often are subtle, can be atypical, and can be mistaken for normal infant movements as well as other seizures and movement disorders. In exceptional cases, ES may manifest solely as brief behavioral arrests with wide-eyed stares, upward eye rolls, or minor head nods. The contractions of ES can be asymmetric, especially in the setting of focal structural lesions. Other seizure types can precede ES or co-occur at onset and within clusters of ES, complicate and delay IESS diagnosis, and therefore delay appropriate first-line treatment. Benign neurologic phenomena, such as an exaggerated startle reflex, shuddering spells, or benign sleep myoclonus, may mimic ES. Abnormal movements in the setting of gastroesophageal reflux are a common misdiagnosis of IESS, albeit a common mimicker. A high index of suspicion is warranted for infants exhibiting unusual “jerks,” “startles,” or “head bobs,” especially if clustering upon awakening, and urgent differential diagnosis of events should follow.
Diagnostic and Etiologic Evaluation
A thorough clinical history and examination should complement an appropriately timed video-EEG for a definitive diagnosis of IESS. Both interictal and ictal electrographic findings can help confirm IESS. Hypsarrhythmia is the most common interictal pattern, characterized by chaotic, high-amplitude, nonepileptiform waves (consistently greater than 200 µV) and intermixed multifocal or generalized epileptiform discharges (Figure 1A). Hypsarrhythmia can be transient and often most prominent in non-REM sleep. Early in the disease course, hypsarrhythmia can be absent or missed in a sizeable minority with verified ES.5 There is poor interrater reliability in the determination of hypsarrhythmia on EEG.6 Interictal abnormalities reliably are present but not specific to IESS. Objective and quantifiable abnormal features (eg, number of spike foci, etc) compiled in scales, such as the BASED score, improve interrater reliability in assessment of the interictal EEG.7 Challenges persist to establish which interictal EEG factors may be most clinically meaningful to the diagnosis, response to treatment, and prediction of relapse in IESS or other relevant epilepsy syndromes.
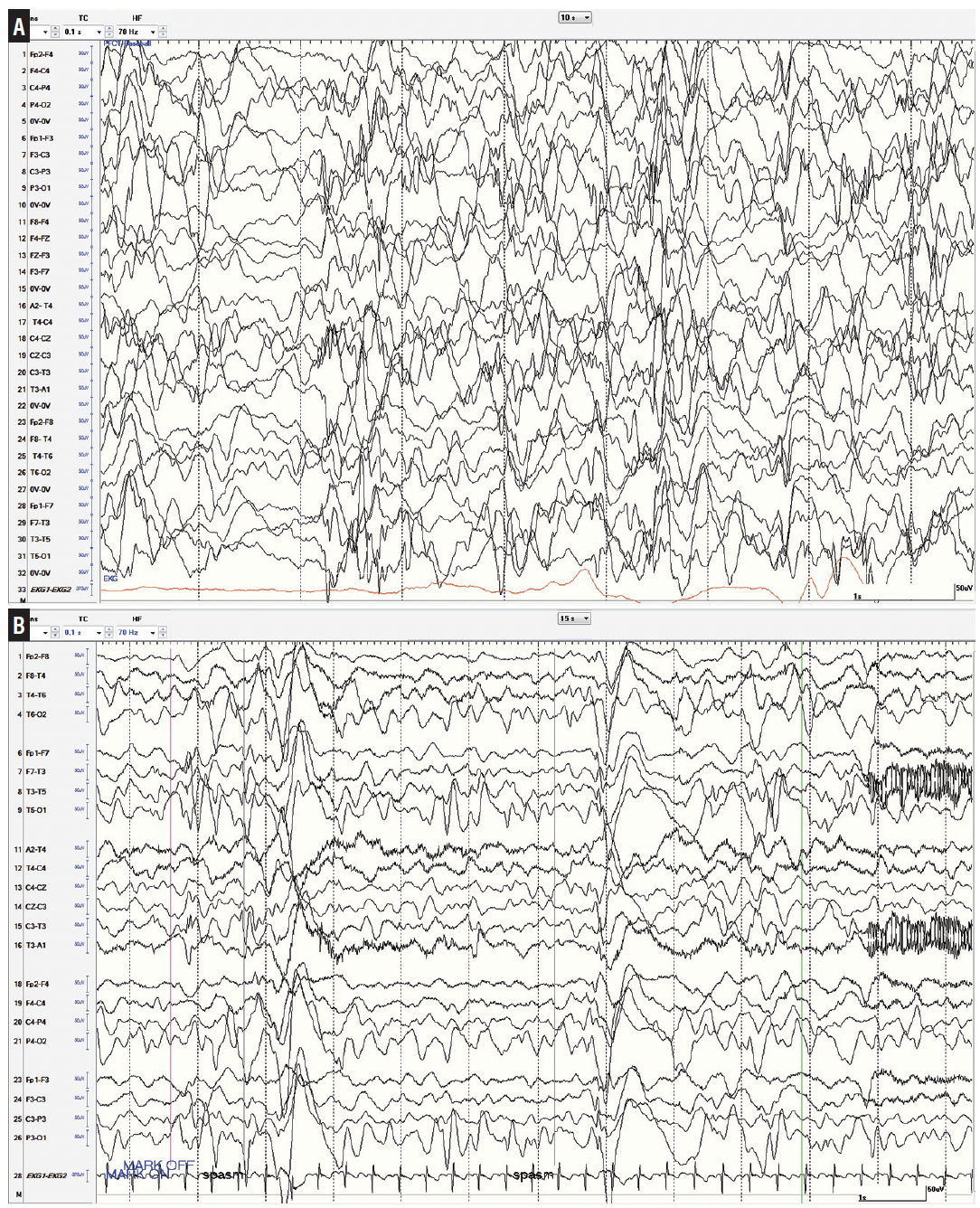
Figure 1. Interictal and ictal features of infantile epileptic spasm syndrome. A, Typical hypsarrhythmia showing chaotic and high-amplitude waves above 200 µV and multifocal epileptiform discharges. B, Epileptic spasm cluster denoted by high-amplitude slow waves with superimposed fast activity leading into an electrodecrement.
ES usually exhibit distinctive ictal EEG features. Most often, a high-amplitude slow transient with superimposed fast activity coincides with the ES and is followed by brief voltage attenuation (Figure 1B). Ictal attenuation may persist for several seconds or longer, sometimes intermixed with pseudonormalization of amplitude throughout the duration of an ES cluster. Other seizure types may precede or cooccur within a cluster of ES. An ictal EEG in an infant consistent with ES is sufficient for diagnosis of IESS, unless a superseding epileptic encephalopathy syndrome with other seizure types including ES is more appropriate, such as Ohtahara syndrome. An ictal EEG is not required for diagnosis of IESS if there is hypsarrhythmia and clinical suspicion for ES. If IESS is suspected and the interictal EEG is not hypsarrhythmic, an ictal EEG is required for diagnosis.4
Establishing an underlying etiology can have important implications for both treatment and prognosis. Numerous and diverse neurologic disorders have been associated with IESS, including genetic without macrostructural abnormalities, structural, infectious, or metabolic causes. The clinical history of events during pregnancy, perinatal, or postnatal periods may be suggestive of a specific etiology. A detailed examination, including a general examination to identify syndromic features, organomegalies, head circumference, and neurocutaneous stigmata, may narrow the differential diagnosis and suggest targeted testing. An MRI should be obtained at diagnosis, and results are abnormal at ES onset in most patients.8 If the etiology remains unclear, if the examination is suggestive of a genetic or chromosomal syndrome, or if there are structural abnormalities with known genetic predispositions, genetic testing should be performed. Multiplex epilepsy gene panels followed by chromosomal microarrays have the highest yield, being diagnostic in >40% of children with IESS without clear etiology after initial evaluation including MRI.9 Metabolic disorders are relatively rarer causes of IESS, but serum, urine, or CSF testing may be warranted if etiology remains unknown. A repeat MRI after 2 years of age when white matter myelination is more complete is warranted in treatment-refractory IESS without a clear cause or when a structural abnormality is suspected, but not detected on initial MRI. Likewise, PET scans can be useful to detect suspected focal structural abnormalities in the presence of a normal-appearing MRI. Overall, an etiology is identified in up to 90% of cases; those without an etiology identified and normal development prior to the onset of IESS may have a better prognosis.10
Disparate neurologic disorders cause IESS with similar convergence of electrographic patterns and developmental impact. Identical neurologic disorders do not equivalently and invariably lead to IESS. Down syndrome is the most common genetic/chromosomal cause of IESS, although other genetic causes have higher rates of IESS. Hypoxic ischemic encephalopathy, intracranial hemorrhage or ischemic infarcts, hypoglycemic encephalopathy, meningitis, and trauma are common prenatal or postnatal, acquired structural causes of IESS. Common developmental structural malformations include focal cortical dysplasia, lissencephaly, and agenesis/dysgenesis of corpus callosum.8,9 The etiology of IESS may affect treatment; it is often a critical determinant of short-term response and long-term developmental outcome.
Treatment
There is consensus that the most effective therapies for children with new-onset IESS include hormonal therapies or vigabatrin. Among the first-line treatments, hormonal therapies are more effective than vigabatrin (except in tuberous sclerosis complex, where vigabatrin remains as first-line therapy) and dual therapy with simultaneous hormonal therapy and vigabatrin may be most efficacious.11 Equally important as the choice and rapid initiation of a first-line treatment regimen is a timely repeat EEG to assess for resolution of ES and normalization of hypsarrhythmic patterns. If remission is not achieved within 1 to 2 weeks, initiation of an alternate standard therapy may optimize chances for remission and consequently improve developmental outcomes.3,12
Dual therapy with high-dose oral corticosteroids and vigabatrin is started simultaneously to treat infants with new-onset IESS at our institution, except in infants at high risk for adverse events with hormonal therapy or in infants for whom specific treatment is indicated (eg, ketogenic diet in pyruvate dehydrogenase deficiency) (Figure 2). An EEG is repeated at 2 weeks to evaluate response; if hypsarrhythmia or ES continue, oral corticosteroids are discontinued and adrenocorticotropin hormone (ACTH) is initiated. If remission is achieved after 2 weeks of either high-dose hormonal therapy, or if both fail sequentially, the treatment is tapered over 2 weeks. Vigabatrin is continued for about 1 year if there was benefit, with the goal of preventing IESS relapse, although there is scant evidence to guide the optimal duration. If remission or benefit is not seen, vigabatrin is discontinued after 4 weeks and second-line medical therapies are given sequentially, with little data to guide the practitioner. Among infants with known or suspected focal structural abnormalities, evaluation for surgical resection is indicated. Among children without clear lateralization on EEG or a resectable MRI lesion, corpus callosotomy may aid in identification of occult focal cortical dysplasia and subsequent resection or result in meaningful improvements in elimination of ES and interictal abnormalities.13
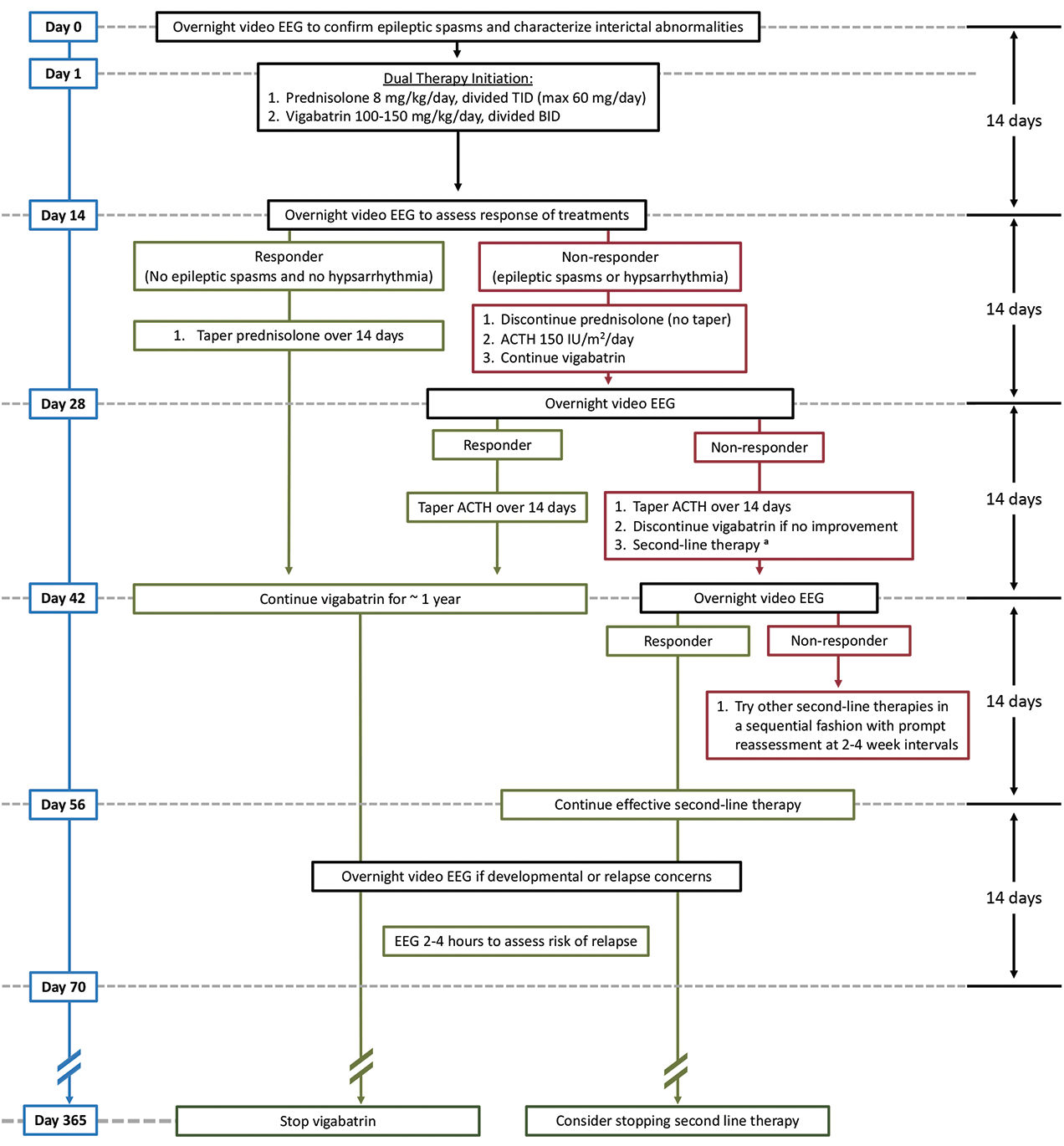
Figure 2. General treatment schema at University of California, Los Angeles Infantile Spasms Program. Dual therapy with simultaneous high-dose prednisolone and vigabatrin is initiated, except when high risk is present for adverse events with hormonal therapy (eg, infection risk or significant cardiac disease) or a specific treatment is otherwise indicated. Nonresponders to prednisolone will switch to adrenocorticotropin hormone. Both hormonal treatments require a 14-day taper.a Second-line treatments with fewer supporting data for efficacy include other antiseizure medications, including benzodiazepines, felbamate, topiramate, and zonisamide. Ketogenic diet therapy may be used and is indicated in some individuals. Pyridoxine-dependent epilepsy may be treated by pyridoxine or leucovorin, and a trial of empiric treatment is warranted in nonresponders.
Adverse effects are monitored with all IESS therapies. Potentially serious adverse effects of hormonal therapy include immunosuppression or infection, hypertension, and hyperglycemia. Adverse effects of vigabatrin include peripheral vision loss and acquired MRI abnormalities. These MRI changes rarely may be reflected by abnormal movements, bradyarrhythmia, or respiratory depression.14
Outcomes and Summary
Long-term outcomes for IESS generally are poor, with high rates of medically refractory epilepsy (especially Lennox-Gastaut syndrome) and intellectual disability. The underlying etiology of IESS is the most important prognostic factor.15 Among infants with unknown etiology and normal development prior to IESS onset, prompt and successful treatment of IESS often yields favorable developmental outcomes.2
1. West WJ. On a peculiar form of infantile convulsions. Lancet. 1841;1:724-725.
2. Kivity S, Lerman P, Ariel R, et al. Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia. 2004;45:255-262.
3. O’Callaghan FJ, Lux AL, Darke K, et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study. Epilepsia. 2011;52:1359-1364.
4. Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63:1349-1397.
5. Demarest ST, Shellhaas RA, Gailliard WD, et al; Pediatric Epilepsy Research Consortium. The impact of hypsarrhythmia on infantile spasms treatment response: observational cohort study from the National Infantile Spasms Consortium. Epilepsia. 2017;58(12):2098-2103.
6. Hussain SA, Kwong G, Millichap JJ, et al. Hypsarrhythmia assessment exhibits poor interrater reliability: a threat to clinical trial validity. Epilepsia. 2015;56(1):77-81.
7. Mytinger JR, Hussain SA, Islam MP, et al. Improving the inter-rater agreement of hypsarrhythmia using a simplified EEG grading scale for children with infantile spasms. Epilepsy Res. 2015;116:93-98.
8. Osborne JP, Lux AL, Edwards SW, et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia. 2010;51(10):2168-2174.
9. Wirrell EC, Shellhaas RA, Joshi C, et al; Pediatric Epilepsy Research Consortium. How should children with West syndrome be efficiently and accurately investigated? Results from the National Infantile Spasms Consortium. Epilepsia. 2015;56(4):617-625.
10. Pellock JM, Hrachovy R, Shinnar S, et al. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51(10):2175-2189.
11. O’Callaghan FJ, Edwards SW, Alber FD, et al; Participating Investigators. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. Lancet Neurol. 2017;16(1):33-42.
12. Yuskaitis CJ, Mytinger JR, Baumer FM, et al. Association of time to clinical remission with sustained resolution in children with new-onset infantile spasms. Neurology. 2022;99(22):e2494-e2503.
13. Baba H, Toda K, Ono T, et al. Surgical and developmental outcomes of corpus callosotomy for West syndrome in patients without MRI lesions. Epilepsia. 2018;59(12):2231-2239.
14. Hussain SA, Tsao J, Li M, et al. Risk of vigabatrin-associated brain abnormalities on MRI in the treatment of infantile spasms is dose-dependent. Epilepsia. 2017;58(4):674-682.
15. Riikonen R. Epidemiological data of West syndrome in Finland. Brain Dev. 2001;23(7):5
Dr. Groves reports no disclosures.
Dr. Hussain has received compensation for research and/or consultancy from Aquestive, Amzell, Eisai, Biopharm Solutions, Eisai, Equilibre, GW Pharma, Marinus, MGC Pharma, Radius, Shennox, UCB Biopharma, Upsher-Smith, West Therapeutic Development, and Zogenix.


