Clinical Presentation
Ms G is age 23 and in nursing school. She has a history of migraine and presents with 1 year of progressive gait and fine motor abnormalities, tremors, and speech changes. At presentation to the neurology clinic, she reports a tendency to arch her back as she walks and says her back and legs feel stiff while ambulating. She has lost her balance twice in the last year. Ms G also has difficulty with fine motor coordination of her hands, which is prominent when she is placing intravenous (IV) lines. She says she is also experiencing a hand tremor, most noticeable when she is holding items. Her speech has become mildly slurred, but she says she has no difficulties with language. Her migraines are unchanged.
On review of systems, Ms G notes an unintentional 10-pound weight loss over the last year, occasional loose stools, and emesis. She has been experiencing some depression, anxiety, and irritability, which have all been greater over the last year compared with her previous baseline. She reports mental fogginess and fatigue but remains independent in her activities of daily living. Ms G does not take any medications and has no family history of neurologic disorders.
Neurologic Examination
On neurologic examination, Ms G was fully oriented and provided a clear history. Her speech was mildly dysarthric and language was intact. She had mild masking of facial expression. Keyser-Fleisher rings were visible on pen-light examination of her eyes, which was later confirmed on slit lamp exam by ophthalmology. Ms G had moderate bradykinesia in all limbs, moderate rigidity throughout, and a mild proximal right upper extremity tremor when the elbow was flexed. Posturing and curling of her fingers was observed on distraction. Ms G was able to stand and ambulate independently, but with tight-appearing trunk and leg muscles that contracted irregularly while walking, leading to a tilting posture.
Diagnostic Studies
A complete blood count (CBC), basic metabolic panel, liver function tests, and 24-hour urinalysis were ordered. Ceruloplasmin, creatinine kinase, and alkaline phosphatase levels were also obtained. As the results in the Table show, Ms G had elevated urine copper and low ceruloplasmin levels.
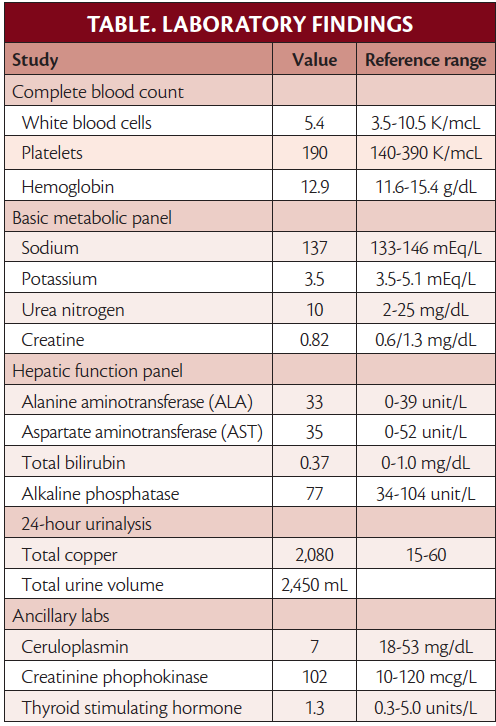
Review of available brain MRI (Figure) demonstrated T2 signal changes in Ms G’s basal ganglia and midbrain.
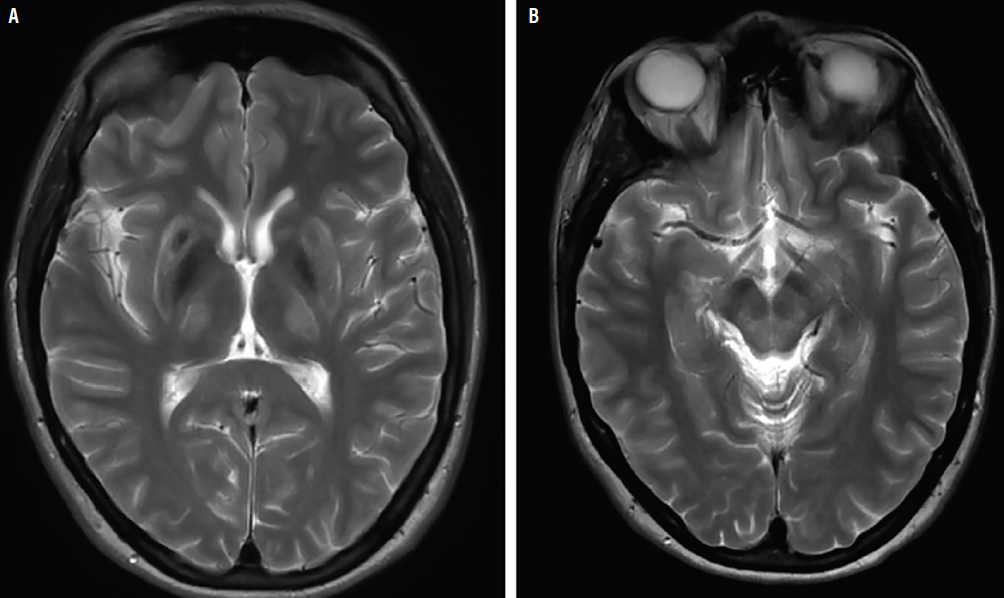
Figure. Hyperintensities in basal ganglia on T2-weighted brain MRI, also known as “face of the giant panda sign.”.
Challenge Questions
1.What genetic abnormality is most likely to be present to explain this condition?
A. Heterozygous Β-glucocerebrosidase mutation
B. Trinucleotide repeats of cytosine-adenine-guanine
C. Compound heterozygous ATP7B mutation
D. HLA-DRB1 variant
E. C282Y or H63D mutations in the HFE gene
Click here for the answer
C: a compound heterozygous mutation in ATPase copper transporting Β (ATP7B) is the most likely genetic abnormality.
2. Which of the following clinical characteristics alone are most suggestive of Wilson disease?
A. Elevated serum copper, low 24-hour urine copper, and acute hepatitis
B. Presence of Kayser-Fleischer rings on slit lamp examination, normal brain MRI, and no neuro-psychiatric symptoms
C. Coombs negative hemolytic anemia, high serum copper, and elevated liver enzymes, with liver copper less than twice the upper limit of normal
D. Elevated serum ceruloplasmin and hyperintense T2 signal in basal ganglia on MRI
E. 24-hour urinary copper more than twice the upper limit of normal, with no acute hepatitis, and serum ceruloplasmin less than10 mg/dL
Click here for the answer
E: 24-hour urinary copper more than twice the upper limit of normal, with no acute hepatitis, and serum ceruloplasmin less than10 mg/dL.
3. Which of the following can be presenting features of neuropsychiatric Wilson disease?
A. Parkinsonism
B. Dystonia
C. Ataxia
D. Tremors
E. Chorea
F. Psychosis
G. All of the above
H. None of the above
Click here for the answer
G: all of the above because phenotypically, Wilson disease can present with variable movement disorders in isolation or combination.
4.What are the most effective treatments available to treat Ms G’s condition?
A. Penicillamine, trientine hydrochloride, and zinc salts
B. Deferoxamine, vitamin C, and phlebotomy
C. Zinc, succimer, dimercaprol, and CaNa2 EDTA
D. EDTA and levodopa
E. Dimercaprol, D-penicillamine, and 2,3 dimercaptopropane-1-sulfonate
Click here for the answer
A: D-penicillamine, trientine hydrochloride, and zinc salts are the 3 pharmacologic modalities available to treat Wilson disease.
Case Resolution
Ms G initiated treatment with 50 mg of zinc acetate 3 times a day and 500 mg of trientine twice daily. These are taken apart from each other and apart from meals. Because Ms G experienced nausea and upset stomach, zinc treatement was discontinued. Improvements in parkinsonism, dystonia, dysphagia, and hypersalivation were seen after 1 month of treatment. Because Ms G continued to experience some truncal dystonia, as described, the anticholinergic trihexyphenidyl was added to her treatment. A liver ultrasound and fibroscan showed mild liver fibrosis. Blood cell counts and liver function tests were followed and remained stable. Ms G had a neuropsychiatric evaluation and began treatment with antidepressants. Genetic testing was performed and confirmed ATP7B mutations in both ATP7B alleles. Counseling and screening were provided to her younger brother.
Discussion
Diagnostic Criteria
Wilson disease should be considered and screened for in anyone under age 50 who has a combination of unexplained neurologic, psychiatric, and/or hepatic dysfunction. The prevalence of Wilson disease varies by geographic location and is estimated to be approximately 1 in 30,000 individuals.2 The mean age of onset for neurologic manifestations of Wilson disease is age 15 to 21, which is later than onset of hepatic symptoms. This can be variable, however, with onset ranging from the first to seventh decades of life.3,4 Neurologic symptoms are the presenting manifestation in 18% to 68% of patients.3 Neurologic symptoms are broad, and common manifestations include dysarthria, dystonia, tremor, ataxia, and parkinsonism. A validated scoring system for the diagnosis of Wilson disease incorporates clinical symptoms, laboratory biochemical data, the presence of Kayser-Fleischer rings on slit-lamp exam, and genetic test results.1
In anyone for whom a diagnosis of Wilson disease is suspected, 24-hour urinary copper levels should be obtained, along with serum ceruloplasmin, serum copper, liver function tests, and a complete blood count. Elevated 24-hour urine copper levels (>40 mcg) and low serum ceruloplasmin (<20 mg/dL) is highly suggestive of Wilson disease. Total serum copper is not a reliable diagnostic in Wilson disease because it includes both ceruloplasmin-bound copper (low in Wilson disease, proportional to low ceruloplasmin) and free copper, which is elevated. The total serum copper level thus may be normal or elevated, particularly if there is acute liver failure, which causes free copper to be released into the serum.5 If biochemical analyses are equivocal, liver biopsy is the diagnostic criterion for copper levels in Wilson disease. Brain MRI in individuals with Wilson disease (Figure) commonly reveals changes on T1- or T2-weighted sequences within the bilateral basal ganglia as well as the thalami and brainstem. A hyperintense T2 signal in the midbrain surrounding the red nuclei may occur in Wilson disease and has been dubbed the “face of the giant panda sign.”6
Genetics of Wilson Disease
Wilson disease is an autosomal recessive genetic disorder that results from mutations in the ATP7B gene, which encodes a P-type ATPase that mediates ATP-dependent transport of copper. ATP7B is highly expressed in hepatocytes where it has dual roles in copper metabolism. Firstly, ATP7B facilitates incorporation of copper into ceruloplasmin, which is then secreted into the bloodstream. Secondly, ATP7B promotes the biliary excretion of copper by sequestering copper into vesicles that are excreted into bile via exocytosis.2 Pathogenic mutations in ATP7B lead to impaired copper excretion, resulting in copper accumulation in hepatocytes with resultant hepatotoxicity and accumulation of excess copper in other organs including the brain. Over 900 pathogenic mutations have been identified in the ATP7B gene to date, with considerable variation in mutation frequency across different geographic cohorts.7 A substantial proportion of individuals with Wilson disease have compound heterozygous mutations, harboring distinct mutations on each copy of the gene.2 In those for whom a pathogenic ATP7B mutation is confirmed, genetic screening should be offered for first-degree family members and can be targeted towards the specific mutations identified in the proband.
Treatment
Treatment of Wilson disease is targeted towards decreasing copper levels by inhibiting copper absorption and promoting copper excretion. Oral zinc preparations interfere with the intestinal absorption of copper, and multiple zinc salt preparations are available. Dosing of zinc is based on the quantity of elemental zinc, with standard dosing of 50 mg of elemental zinc 3 times daily. Copper chelation therapies include trientine and D-penicillamine, which both increase urinary copper excretion. In a subset of individuals, these copper chelation treatments may paradoxically lead to worsening of neurologic symptoms, proposed to be secondary to transient copper level elevation in the brain as existing copper stores are mobilized.8 Trientine and D-penicillamine have comparable rates of neurologic worsening but trientine is better tolerated in regards to adverse side effects.9
Future Directions
Potential treatments under investigation include ammonium tetrathiomolybdate, which interferes with intestinal copper absorption when administered with food and also forms complexes with copper and albumin in the bloodstream, preventing cellular uptake and promoting biliary excretion of copper.10 A randomized controlled trial comparing trientine to ammonium tetrathiomolybdate suggested lower rates of paradoxical neurologic worsening in participants treated with tetrathiomolybdate.11 A recent phase 2 clinical trial of tetrathiomolybdate showed a significant reduction in free copper in people with Wilson disease treated with tetrathiomolybdate, with no cases of paradoxical neurologic worsening.12
1. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139-142.
2. Chang IJ, Hahn SH. The genetics of Wilson disease. Handb Clin Neurol. 2017;142:19-34.
3. Lorincz MT. Neurologic Wilson’s disease. Ann N Y Acad Sci. 2010;1184:173-187.
4. Ala A, Borjigin J, Rochwarger A, Schilsky M. Wilson disease in septuagenarian siblings: raising the bar for diagnosis. Hepatology. 2005;41(3):668-670.
5. Patil M, Sheth KA, Krishnamurthy AC, Devarbhavi H. A review and current perspective on Wilson disease. J Clin Exp Hepatol. 2013;3(4):321-336.
6. Prashanth LK, Sinha S, Taly AB, Vasudev MK. Do MRI features distinguish Wilson disease from other early onset extrapyramidal disorders? An analysis of 100 cases. Mov Disord. 2010;25(6):672-678.
7. Shribman S, Poujois A, Bandmann O, Czlonkowska A, Warner TT. Wilson’s disease: update on pathogenesis, biomarkers and treatments. J Neurol Neurosurg Psychiatry. 2021.
8. Litwin T, Dziezyc K, Karlinski M, Chabik G, Czepiel W, Czlonkowska A. Early neurological worsening in patients with Wilson disease. J Neurol Sci. 2015;355(1-2):162-167.
9. Weiss KH, Thurik F, Gotthardt DN, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11(8):1028-1035 e1021-1022.
10. Brewer GJ, Dick RD, Yuzbasiyan-Gurkin V, Tankanow R, Young AB, Kluin KJ. Initial therapy of patients with Wilson disease with tetrathiomolybdate. Arch Neurol. 1991;48(1):42-47.
11. Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63(4):521-527.
12. Weiss KH, Askari FK, Czlonkowska A, et al. Bis-choline tetrathiomolybdate in patients with Wilson disease: an open-label, multicentre, phase 2 study. Lancet Gastroenterol Hepatol. 2017;2(12):869-876.
SB and SM report no disclosures
DB has served as a consultant and site investigator for Alexion Pharmaceuticals and Ultragenyx Pharmaceuticals



