Definition
Medulloblastoma is defined by the World Health Organization (WHO) as "an embryonal neuroepithelial tumor arising in the cerebellum or dorsal brainstem, presenting mainly in childhood and consisting of densely packed small round undifferentiated cells with mild to moderate nuclear pleomorphism and high mitotic count." [1] These tumors display considerable clinical and biologic heterogeneity, and they have been further defined molecularly and histologically.
The major molecular groups include WNT-activated medulloblastomas (10% of cases), Sonic hedgehog (SHH)-activated (30%), and non-WNT/non-SHH tumors that include "group 3" (25%) and "group 4" (35%). Additional molecular subgroups within these major groups are anticipated (please see Molecular/Genetics). Histologic subtypes of medulloblastoma include: (1) desmoplastic/nodular type, (2) medulloblastoma with extensive nodularity, and (3) large-cell/anaplastic medulloblastoma. In addition to locally aggressive behavior with fourth ventricular obstruction and hydrocephalus, medulloblastomas can metastasize via cerebrospinal fluid (CSF) pathways.
Although the etiology and pathogenesis of medulloblastoma is not entirely understood at present, some recent molecular genetic studies have provided important insights into possible disease mechanisms. [2] (See Molecular/Genetics.)
Epidemiology
Medulloblastoma is the most common malignant central nervous system (CNS) tumor of childhood, with an annual incidence of about 0.5-0.8/100,000 in children younger than 19 years. [3] The vast majority of medulloblastomas occur before age 16 years, and there is a bimodal peak of incidence between ages 3-4 years and 8-9 years. About 10% of cases arise in infants. Males are affected more frequently than females, with a ratio of about 2:1.
Dissemination of medulloblastoma within cerebrospinal fluid (CSF) pathways is a defining pathobiologic characteristic of this tumor, and about 30% of patients will have CSF metastasis at presentation. [4] Adult cases are unusual, accounting for less than 1% of brain tumors. Medulloblastoma is rare beyond the fifth decade.
Location
The majority of medulloblastomas arise from the inferior cerebellar vermis, from which they extend into and typically fill the fourth ventricle. Obstruction of flow of cerebrospinal fluid (CSF) will produce hydrocephalus above this level. The neoplasm can also invade adjacent brainstem structures, including the cardiorespiratory centers of the fourth ventricular floor. The previously mentioned tendency of medulloblastoma to spread via CSF pathways can lead to diffuse "sugar coating" of the subarachnoid space and to nodular growths along the spinal cord or even ventricular surfaces. A smaller proportion of medulloblastomas occur in one of the cerebellar hemispheres of patients who are typically older (adolescents or young adults), a subset in which the desmoplastic/nodular variant predominates. [2]
Clinical Features and Imaging
Symptoms and signs of an expanding posterior fossa mass may include nausea and vomiting, headaches, and ataxia. [2, 4] Hydrocephalus and increased intracranial pressure may result in papilledema. The development of cerebellar tonsillar herniation may be accompanied by neck pain. Meningeal irritation due to spread of tumor in the subarachnoid space may lead to neck stiffness and head tilt. Spinal metastases may result in focal findings at a spinal cord level or nerve root pain. Infants with medulloblastoma may present with failure to thrive, vomiting, and irritability.
Medulloblastomas appear as hyperdense, noncalcified lesions of the fourth ventricle or cerebellar hemisphere on computed tomography (CT) scans. Magnetic resonance imaging (MRI) typically reveals a solid fourth ventricular mass that is hypointense on the T1-weighted image, hyperintense on T2-weighted images, and brightly enhancing on T1-weighted images after contrast administration (see the images below).
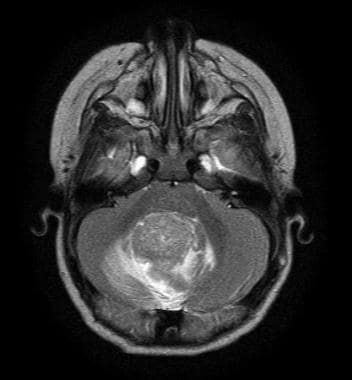 Medulloblastoma Pathology. This axial T2 magnetic resonance image shows a cerebellar vermian midline mass with contrast enhancement and obstruction of the fourth ventricle.
Medulloblastoma Pathology. This axial T2 magnetic resonance image shows a cerebellar vermian midline mass with contrast enhancement and obstruction of the fourth ventricle.
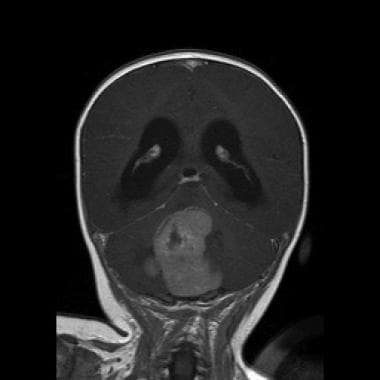 Medulloblastoma Pathology. The coronal T1 magnetic resonance image shows a cerebellar vermian midline mass with contrast enhancement and obstruction of the fourth ventricle.
Medulloblastoma Pathology. The coronal T1 magnetic resonance image shows a cerebellar vermian midline mass with contrast enhancement and obstruction of the fourth ventricle.
Cerebellar hemispheric tumors of older children and adults show similar imaging features, except that contrast enhancement is more variable than that in young children. The "medulloblastoma with extensive nodularity" is a subtype that occurs in young children and has a distinctly lobular appearance on imaging, as shown in the following image.
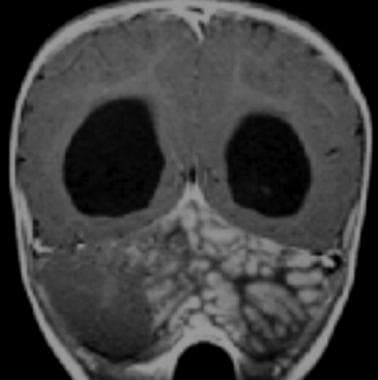 Medulloblastoma Pathology. A lateral cerebellar mass with a "grapelike" lobular appearance that is characteristic of the extensively nodular medulloblastoma is revealed in this magnetic resonance image.
Medulloblastoma Pathology. A lateral cerebellar mass with a "grapelike" lobular appearance that is characteristic of the extensively nodular medulloblastoma is revealed in this magnetic resonance image.
They typically show restriction diffusion on apparent diffusion coefficient (ADC) maps that are consistent with high cellularity (see the image below).
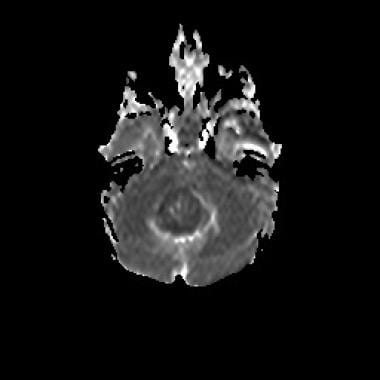 Medulloblastoma Pathology. An apparent diffusion coefficient (ADC) map of a histologically confirmed medulloblastoma shows restricted diffusion consistent with a tumor of high cellularity. The differential diagnosis will include anaplastic ependymoma, atypical teratoid rhabdoid tumor (ATRT), or other forms of highly proliferative malignant neuroectodermal tumors.
Medulloblastoma Pathology. An apparent diffusion coefficient (ADC) map of a histologically confirmed medulloblastoma shows restricted diffusion consistent with a tumor of high cellularity. The differential diagnosis will include anaplastic ependymoma, atypical teratoid rhabdoid tumor (ATRT), or other forms of highly proliferative malignant neuroectodermal tumors.
Mass spectroscopy reveals elevation of choline but little, if any, N-acetyl aspartate (NAA) peak. Elevation of the taurine peak may also be seen.
Gross Findings
Medulloblastomas are typically soft, fleshy, gray-tan tumors with variable hemorrhage and sometimes necrosis. Extensive stromal reticulin deposition may impart a firm consistency to the desmoplastic variant. Infiltration of the meninges and subarachnoid space may be apparent, as well as frank invasion of adjacent structures.
Microscopic Findings
All medulloblastomas are considered to be malignant and invasive tumors; they are thus classified as grade 4 by the World Health Organization (WHO). [1] These masses are highly cellular neoplasms composed of cells with small- to medium-sized, hyperchromatic nuclei and little apparent cytoplasm, as seen in the image below.
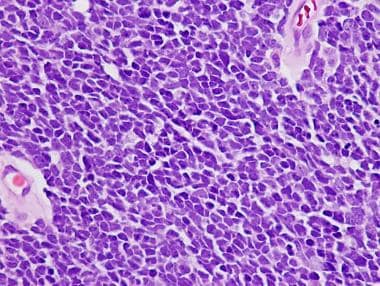 Medulloblastoma Pathology. This example of a classic medulloblastoma shows a diffuse pattern of tumor growth with poor cellular differentiation, nuclear molding, and minimal indistinct cytoplasm.
Medulloblastoma Pathology. This example of a classic medulloblastoma shows a diffuse pattern of tumor growth with poor cellular differentiation, nuclear molding, and minimal indistinct cytoplasm.
Nuclear pleomorphism may be present to some extent, but it is usually not marked. Molding of adjacent cell nuclei may be marked due to high cell density. Nucleoli are not typically prominent, except in the large-cell/anaplastic variant. Mitoses are usually plentiful, as is the necrosis (apoptosis) of individual cells in the form of nuclear pyknosis, fragmentation, or karyorrhexis. Necrosis may be present, and although pseudopalisading necrosis and vascular endothelial proliferation are uncommon, they may occur. Homer Wright ("neuroblastic") rosettes may be found in about 40% of cases (see the image below), but such structures may be subtle or absent, and they are not required for the diagnosis. Nodular foci of tumor may present a less cellular, more "differentiated" character that rarely may include mature-appearing ganglion cells.
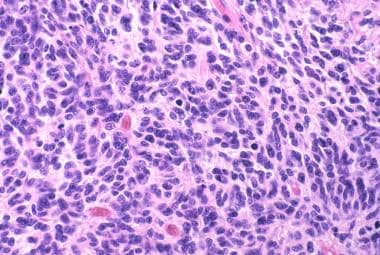 Medulloblastoma Pathology. This high-power microscopy view of a classic medulloblastoma demonstrates nuclear molding and mitotic activity.
Medulloblastoma Pathology. This high-power microscopy view of a classic medulloblastoma demonstrates nuclear molding and mitotic activity.
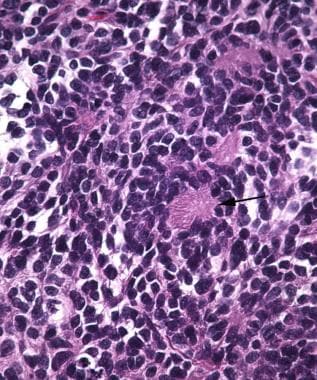 Medulloblastoma Pathology. Homer-Wright rosettes (arrow) are characteristically seen in medulloblastoma and other tumors with neuroblastic differentiation.
Medulloblastoma Pathology. Homer-Wright rosettes (arrow) are characteristically seen in medulloblastoma and other tumors with neuroblastic differentiation.
The term "small blue cell tumor" is somewhat of a misnomer, because the neoplastic nuclei of medulloblastomas are usually substantially larger than those of native cerebellar granule cell neurons. This point becomes relevant in the proper interpretation of intraoperative touch preparations or frozen-section material obtained from posterior fossa biopsies (see the image below).
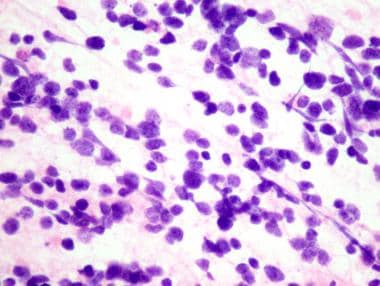 Medulloblastoma Pathology. The intraoperative cytopreparation shows a medulloblastoma with poorly differentiated cells that have little cytoplasm and nuclear streaming resulting from crush artifact.
Medulloblastoma Pathology. The intraoperative cytopreparation shows a medulloblastoma with poorly differentiated cells that have little cytoplasm and nuclear streaming resulting from crush artifact.
Native granule cells have small, round, hyperchromatic nuclei that are about the size of a mature lymphocyte nucleus (7 microns). Correlation of histology with imaging findings that provide the location and character of the lesion (solid vs cystic vs mural nodule) will greatly aid diagnostic interpretation.
Although the majority of medulloblastomas have the classic histopathology described above, several variants have been defined; they have distinct clinical, as well as microscopic, characteristics. It should be noted that the usual type (classic) medulloblastomas may contain foci of increased anaplasia, suggesting progressive transformation, that should be documented in the pathology report. Minor foci of pale nodules may not be of clinical significance. However, the variants described below are those in which features of nodularity or a large-cell/anaplastic phenotype are the predominant histopathology.
The desmoplastic/nodular medulloblastoma is a histologic variant that usually arises in a cerebellar hemisphere and is defined by the presence of a reticulin-rich stroma and reticulin-poor, nodular, pale islands with expression of markers of neuronal differentiation (see the images below).
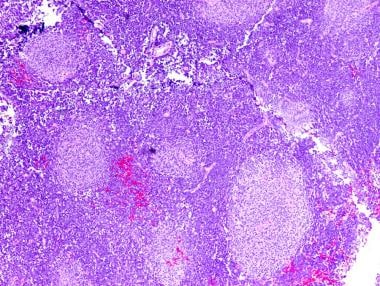 Medulloblastoma Pathology. Nodular (desmoplastic) medulloblastoma with pale nodules of differentiating neuroblasts (neurocytes). Note the abundant intervening less-differentiated internodular region.
Medulloblastoma Pathology. Nodular (desmoplastic) medulloblastoma with pale nodules of differentiating neuroblasts (neurocytes). Note the abundant intervening less-differentiated internodular region.
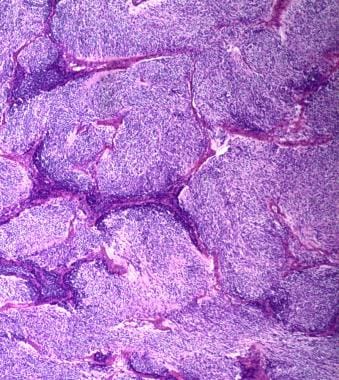 Medulloblastoma Pathology. Extensively nodular medulloblastoma with minimal or no internodular component is depicted.
Medulloblastoma Pathology. Extensively nodular medulloblastoma with minimal or no internodular component is depicted.
Tumor within reticulin-rich areas is usually highly cellular and proliferatively active, whereas the reticulin-poor nodular foci show less mitotic activity and more neuronal differentiation. Evidence exists that such tumors have distinctive molecular characteristics (discussed below). An "extensively nodular" form of medulloblastoma may arise in children younger than 3 years. [1, 5] Such tumors have occasionally been noted to undergo neurocytic or gangliocytic maturation after chemotherapy and/or radiation.
A large cell/anaplastic medulloblastoma is composed of large neoplastic cells with vesicular nuclei, prominent nucleoli, and histologic anaplasia. (see the following image). [1] This histologically defined entity accounts for approximately 10% of all medulloblastomas and can be demonstrated to exhibit neuronal differentiation by immunohistochemical detection of neuronal lineage antigens such as synaptophysin. It may be distinguished from the atypical teratoid/rhabdoid tumor (AT/RT) by its strong expression of INI-1 (BAF47); AT/RT is typically negative for this antigen. The large cell/anaplastic medulloblastoma is a more aggressive variant and is less responsive to standard therapies.
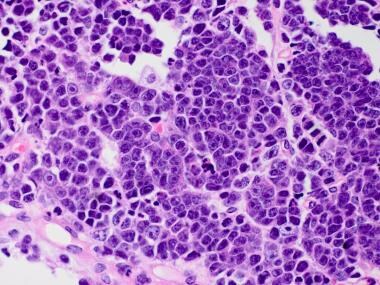 Medulloblastoma Pathology. This example of a large cell medulloblastoma has large vesicular nuclei, distinct nucleoli, and a vague resemblance to large cell lymphoma.
Medulloblastoma Pathology. This example of a large cell medulloblastoma has large vesicular nuclei, distinct nucleoli, and a vague resemblance to large cell lymphoma.
Anaplasia of medulloblastomas has been defined as having an increased nuclear size, with more striking nuclear pleomorphism and molding than the classic type; high mitotic activity with atypical forms; frequent apoptotic bodies; and a distinctive wrapping of one tumor cell around another (see the images below).
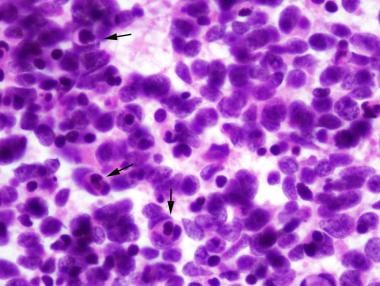 Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma shows significant cytologic anaplasia, frequent cell wrapping (arrows), and apoptotic bodies in intraoperative cytopreparation.
Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma shows significant cytologic anaplasia, frequent cell wrapping (arrows), and apoptotic bodies in intraoperative cytopreparation.
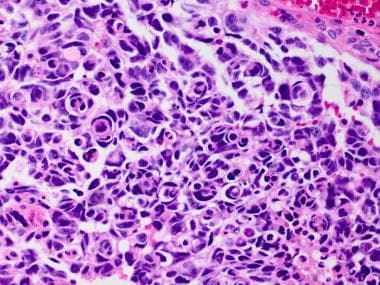 Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma shows significant cytologic anaplasia, frequent cell wrapping, and apoptotic bodies.
Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma shows significant cytologic anaplasia, frequent cell wrapping, and apoptotic bodies.
Variable degrees of anaplasia may be encountered in even classic medulloblastomas such that a more refined grading system coupled with tumor molecular characteristics will allow for more precise patient stratification for treatment. Histologic transformation of more typical types of medulloblastoma to an anaplastic type is well documented.
Rare medulloblastomas may show evidence of skeletal muscle (medullomyoblastoma) or melanocytic differentiation. Data on the natural history of these tumors suggest a similar biologic behavior as that of more typical medulloblastoma types.
Immunohistochemistry
The central nervous system (CNS) origins of medulloblastoma can be confirmed by immunohistochemical studies. Synaptophysin, neuron-specific enolase, microtubule associated protein-2 (MAP-2), and class-III beta tubulin will be at least focally immunoreactive in most of these primitive neuroectodermal tumors (see the image below).
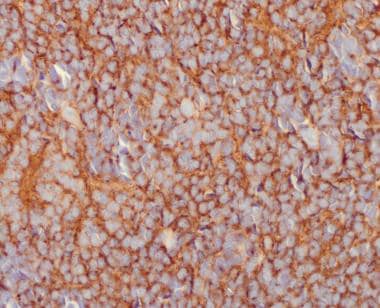 Medulloblastoma Pathology. Immunopositivity for synaptophysin in a medulloblastoma is noted.
Medulloblastoma Pathology. Immunopositivity for synaptophysin in a medulloblastoma is noted.
Reticulin-free nodules of the desmoplastic variant are typically reactive for markers of neuronal lineage. Vimentin is usually reactive although quite nonspecific. Variable expression for neurofilament proteins has been documented, and immunoreactivity is dependent on the neurofilament subtype and the antibody used.
Glial fibrillary acidic protein (GFAP) is most often positive in cells with fibrillary processes that appear to represent reactive (intratumoral) astrocytes. Occasional examples of medulloblastoma (estimated at around 10% of these tumors) will show distinct GFAP-immunoreactivity of the perinuclear cytoplasm. Rare examples of medulloblastoma may contain spindle cells or, occasionally, "strap" cells with cross-striations consistent with myogenic differentiation and, accordingly, show strong reactivity for desmin (and other myogenic markers).
Molecular/Genetics
The most common cytogenetic change found in medulloblastomas (over 40%) is loss of chromosome 17p, which is frequently accompanied by duplication of the long arm, resulting in an isochromosome 17q. One of the candidate tumor suppressor genes on 17p is KCTD11, which inhibits a Sonic hedgehog (SHH) signaling pathway known to regulate cerebellar granule cell proliferation during development. [6] During normal brain development, SHH protein secreted from Purkinje cells inhibits a cell-surface receptor (encoded by the Patched [PTCH] gene on chromosome 9), thus promoting proliferation of cerebellar external granule layer (EGL) cells. The EGL cells give rise to internal granule cells, which are the most abundant neurons of the brain.
Patients with the nevoid basal cell carcinoma syndrome (NBCCS) (Gorlin syndrome) have germline mutations of PTCH, a tumor suppressor gene. When active, PTCH inhibits Smoothened (SMO), a cell surface protein that regulates a cell proliferation pathway, including several intracellular downstream effectors such as GLI1 and MYCN. Inactivating PTCH mutations in patients with nevoid basal cell carcinoma syndrome (NBCCS) presumably releases susceptible cells from PTCH -mediated inhibition of cell proliferation, with resulting neoplasia.
PTCH mutations (allelic losses) have been identified in about 10-18% of medulloblastomas, especially the desmoplastic type, but such mutations are much less common in the most common classic variant. Copy gain and aberrant expression of FOXG1, a downstream effector of SHH signaling with a putative role for maintaining a persistent undifferentiated state in neuroepithelial stem cells, has also been reported to be a frequent event in medulloblastoma.
Some patients with germline mutations of the adenomatous polyposis (APC) gene may develop medulloblastoma in addition to their predisposition to colon cancer (familial adenomatous polyposis [FAP]). The APC protein is an inhibitor of the WNT pathway. Turcot syndrome describes the subset of patients with FAP that develop medulloblastoma.
More recently, there has been emerging evidence that neural stem or progenitor cells may give rise to embryonal tumors of the nervous system. [7]
Based on combined data emerging from advanced molecular characterization and correlation with clinical outcome, medulloblastomas have been divided into four clinically relevant groups, depending on the activated signaling pathway or other molecular features of a given tumor:
-
The WNT-activated group accounts for about 10% of medulloblastomas, arises in older children and occasionally in adults, and carries a good prognosis. Mutations of CTNNB1 and DDX3X are frequent in this type with germline mutations of APC occasionally observed.
-
Sonic hedgehog (SHH) pathway activation occurs in about 30% of medulloblastomas, may affect infants as well as older children and adults, have PTCH/SMO/SUFU mutations, and may show a desmoplastic/nodular histology. This group has also been divided into four clinically relevant subgroups (SHH-1 to SHH-4). For example, an SHH-activated, TP53-mutant medulloblastoma (SHH-3) may show large cell anaplastic histology and has a worse prognosis than TP53-wildtype SHH-activated tumors (SHH-1, -2, and -4).
-
Two other molecular groups of non-WNT/non-SHH medulloblastomas have been designated " group 3 " ( 25% of cases) and " group 4 " (35% of cases). Such tumors are more genetically heterogeneous and often show i(17q), 1X, -18, and MYC amplification among other alterations. Tumors in group 3 tend to be biologically aggressive, have MYC or MYCN amplifications, and large cell/anaplastic histology. At least eight subgroups of the non-WNT/non-SHH medulloblastomas have been identified at the time of this updated chapter. [8]
Boulay et al characterized gene regulation mechanisms in group 3 medulloblastoma tumors through genome-wide chromatin and expression profiling. [9] They found that transcription factor OTX2 occupies the most active distal sites in these tumors. Using chromatin profiling, they found that OTX2, together with transcription factor NEUROD1, controls the group 3 medulloblastoma active enhancer landscape. [9]
Laneve et al showed that linc-NeD125 is significantly overexpressed in group 4 medulloblastomas. [10] In their study, linc-NeD125 was able to recruit the microRNA (miRNA)-induced silencing complex (miRISC) and to directly bind the microRNAs miR-19a-3p, miR-19b-3p, and miR-106a-5p. Linc-NeD125 acted as a competing endogenous RNA that led to de-repression of their targets (which are group 4 medulloblastoma driver genes), and linc-NeD125 downregulation reduced the proliferation of group 4 cells. [10]
Shih et al attempted to determine whether subgroup affiliation and cytogenetic biomarkers could support or supplant clinical variables for prognostication in patients with medulloblastoma. [11] They concluded that combining subgroup and cytogenetic biomarkers with established clinical biomarkers substantially improves patient prognostication, even in the context of heterogeneous clinical therapies. The prognostic significance of most molecular biomarkers is restricted to a specific subgroup. The authors identified a small panel of cytogenetic biomarkers that reliably identifies very high-risk and very low-risk groups of patients. These biomarkers are an excellent tool for selecting patients for therapy intensification and therapy de-escalation in future clinical trials. [11]
Tumor Spread and Staging
Intraaxial tumor spread along cerebrospinal fluid (CSF) pathways is characteristic of medulloblastoma; it is identified in about 30% of patients at presentation. Subpial and parenchymal infiltration of tumor cells is not uncommon. Distant metastases are exceptional. Occasionally, tumors have metastasized outside of the nervous system via ventriculoperitoneal shunts or other iatrogenic means.
Patients are currently staged (or stratified) into the following risk groups. Average risk is defined as the following:
-
Age older than 3 years but less than 22 years at presentation
-
No or minimal residual tumor (< 1.5 cm maximum diameter) by postoperative imaging
-
No evidence of metastatic spread of disease within the neuraxis. High-risk patients will have significant residual disease after surgery (>1.5 cm maximum diameter) and/or evidence of spinal or other metastasis (usually within the neuraxis). Patients younger than 3 years are also considered high risk, with the exception of those very young patients who have the extensively nodular histologic variant, in which a better prognosis has been observed.
Patients who do survive often experience significant neurologic impairment due to unavoidable side effects of radiotherapy. Current research is focused on identifying biomarkers of disease that will allow for better risk assessment as well as more refined treatments that are directed to individual tumor types.
Prognosis and Predictive Factors
Treatment for medulloblastoma typically includes surgery (with gross total excision, if possible), craniospinal radiation therapy, and adjuvant chemotherapy. [4] The overall 5-year survival rate is currently around 60%.
The expression of nuclear beta-catenin occurs in 15-20% of medulloblastomas; it is considered to correlate with activation of the WNT signaling pathway and a better patient prognosis. In contrast, the MYCC gene is amplified in around 10% of medulloblastomas; it is most often observed in anaplastic variants of medulloblastoma (see the image below).
Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma shows significant cytologic anaplasia, frequent cell wrapping (arrows), and apoptotic bodies in intraoperative cytopreparation.
Studies suggest that MYCC alterations may be involved in the progression of medulloblastoma to anaplastic variants. Other molecular alterations associated with a poor prognosis include chromosome 1q gain, MYCN amplification (see the following image), C-ERBB2 overexpression, and p53 mutation/expression, whereas loss of chromosome 6 and TRK-C expression are related to a better prognosis. Ongoing and future studies of medulloblastoma biology may provide a better understanding of tumorigenesis that will lead to more accurate stratification of patients for therapy and novel therapeutic targets.
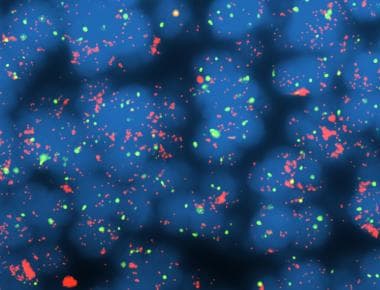 Medulloblastoma Pathology. MYCC amplification is a poor prognostic marker in medulloblastoma, and it is often associated with the anaplastic/large cell phenotype.
Medulloblastoma Pathology. MYCC amplification is a poor prognostic marker in medulloblastoma, and it is often associated with the anaplastic/large cell phenotype.
Differential Diagnosis
The following conditions should be considered in the differential diagnosis of medulloblastoma:
The most important differential diagnosis in children is an ependymoma arising in the fourth ventricle, which can present a similar gross appearance to that of medulloblastoma. Although cerebellar pilocytic astrocytomas tend to present as a cystic structure with an enhancing mural nodule, an occasional solid tumor of this type may mimic the imaging appearance of a medulloblastoma. The large-cell/anaplastic variants may be distinguished from atypical teratoid/rhabdoid tumor (AT/RT) by imumnostaining for INI1, which is positive in medulloblastoma but negative in AT/RT.
-
Medulloblastoma Pathology. This axial T2 magnetic resonance image shows a cerebellar vermian midline mass with contrast enhancement and obstruction of the fourth ventricle.
-
Medulloblastoma Pathology. The coronal T1 magnetic resonance image shows a cerebellar vermian midline mass with contrast enhancement and obstruction of the fourth ventricle.
-
Medulloblastoma Pathology. A lateral cerebellar mass with a "grapelike" lobular appearance that is characteristic of the extensively nodular medulloblastoma is revealed in this magnetic resonance image.
-
Medulloblastoma Pathology. An apparent diffusion coefficient (ADC) map of a histologically confirmed medulloblastoma shows restricted diffusion consistent with a tumor of high cellularity. The differential diagnosis will include anaplastic ependymoma, atypical teratoid rhabdoid tumor (ATRT), or other forms of highly proliferative malignant neuroectodermal tumors.
-
Medulloblastoma Pathology. This example of a classic medulloblastoma shows a diffuse pattern of tumor growth with poor cellular differentiation, nuclear molding, and minimal indistinct cytoplasm.
-
Medulloblastoma Pathology. Homer-Wright rosettes (arrow) are characteristically seen in medulloblastoma and other tumors with neuroblastic differentiation.
-
Medulloblastoma Pathology. The intraoperative cytopreparation shows a medulloblastoma with poorly differentiated cells that have little cytoplasm and nuclear streaming resulting from crush artifact.
-
Medulloblastoma Pathology. Nodular (desmoplastic) medulloblastoma with pale nodules of differentiating neuroblasts (neurocytes). Note the abundant intervening less-differentiated internodular region.
-
Medulloblastoma Pathology. Extensively nodular medulloblastoma with minimal or no internodular component is depicted.
-
Medulloblastoma Pathology. Large cell medulloblastoma with large vesicular nuclei, distinct nucleoli, and a vague resemblance to large cell lymphoma is shown. Note a "starry sky" appearance in this tumor.
-
Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma shows significant cytologic anaplasia, frequent cell wrapping (arrows), and apoptotic bodies in intraoperative cytopreparation.
-
Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma reveals significant cytologic anaplasia, frequent cell wrapping (arrow), and apoptotic bodies.
-
Medulloblastoma Pathology. Immunopositivity for synaptophysin in a medulloblastoma is demonstrated.
-
Medulloblastoma Pathology. MYCC amplification is a poor prognostic marker in medulloblastoma, and it is often associated with the anaplastic/large cell phenotype.
-
Medulloblastoma Pathology. This high-power microscopy view of a classic medulloblastoma demonstrates nuclear molding and mitotic activity.
-
Medulloblastoma Pathology. This example of a large cell medulloblastoma has large vesicular nuclei, distinct nucleoli, and a vague resemblance to large cell lymphoma.
-
Medulloblastoma Pathology. This histologic sample of anaplastic medulloblastoma shows significant cytologic anaplasia, frequent cell wrapping, and apoptotic bodies.
-
Medulloblastoma Pathology. Immunopositivity for synaptophysin in a medulloblastoma is noted.









